Las vasculitis se definen como un grupo de enfermedades y síndromes que cursan con inflamación de los vasos sanguíneos -arterias, arteriolas, capilares, vénulas y venas-, que ocasionan una disminución del flujo vascular o incluso una interrupción completa del mismo, produciendo una gran heterogeneidad de manifestaciones clínicas en función de los distintos órganos afectados y grado de compromiso de los mismos. Pueden ocurrir como una enfermedad primaria o ser secundarias a otra enfermedad subyacente, y por otro lado pueden limitarse sólo a la piel ?vasculitis cutánea- o afectar simultáneamente a varios órganos y aparatos -vasculitis sistémicas. Su tratamiento y pronóstico resultan igualmente muy heterogéneo, en función los órganos implicados y el grado de afectación de los mismos, siendo con frecuencia enfermedades graves, a veces incluso fatales, que requieren un rápido reconocimiento y tratamiento. En este post se realiza una revisión general de las vasculitis, enfocada a a aquellos aspectos teórico prácticos que pueden ser de mayor utilidad al médico de familia.CLASIFICACIÓN
No existe una clasificación unánimemente aceptada de las vasculitis. Las clasificaciones más utilizadas se basan en el tamaño de los vasos sanguíneos afectados (tabla 1), el mecanismo patogénico implicado -producción anticuerpos, depósitos de inmunocomplejos-, el proceso inflamatorio que originan -lesión necrosante o granulomatosa- y en base a si son son primarias o secundarias.
Tabla 1. Clasificación de la vasculitis según el tamaño de los vasos afectados.
Vasculitis de vasos grandes
Arteritis de células gigantes. (Arteritis de la temporal).
Arteritis de Takayasu.
Vasculitis de vasos medianos
Poliarteritis nodosa (PAN).
Enfermedad de Kawasaki.
Vasculitis primaria del sistema nervioso central.
Vasculitis de vasos pequeños
Causadas por ANCA (Anticuerpos anticitoplasma de neutrófilos):
Poliarteritis microscópica.
Granulomatosis de Wegener.
Vasculitis granulomatosa eosinofilica. (Síndrome de Churg-Strauss).
Vasculitis por hipersensibilidad.
Vasculitis por IgA (Síndrome de Schönlein-Henoch).
Urticaria vasculitis hipocomplementémica.
Vasculitis crioglobulinémica.
Vasculitis leucocitoclástica cutánea.
Vasculitis secundarias a enfermedades del tejido conectivo: Lupus eritematoso sistémico, artritis reumatoide , policondritis recidivante , enfermedad de Behçet , etc.
Vasculitis secundarias a infecciones virales: Virus de hepatitis B y C, VIH, citomegalovirus, virus de Epstein - Barr, y parvovirus B19.
Vasculitis paraneoplásicas: Secundarias a neoplasias linfoproliferativas, mieloproliferativas y carcinomas.
EPIDEMIOLOGÍA
Las vasculitis son enfermedades infrecuentes; se estima una incidencia 10-20 casos por millón de habitante, siendo mas comunes en hombres y aumentado su incidencia con la edad. Las vasculitis por hipersensibilidad son las más frecuentes, seguida en segundo lugar de la arteritis de células gigantes.
ETIOPATOGÉNIA
La inflamación de los vasos puede producirse por tres mecanismos:
A) Lesión directa del agente sobre el vaso: Se han descrito en relación con agentes infecciosos (virales o bacterianos), embolías de colesterol e inyección de ciertos materiales tóxicos (ejem. drogas). Este es el mecanismo etiopatogénico menos frecuente implicado en el desarrollo de las vasculitis.
B) Inflamación desencadenada por anticuerpos dirigidos contra algún componente de la pared vascular: se han identificado anticuerpos antimembrana basal de riñon y pulmon y anticuerpos anti células endoteliales que inician el proceso inflmatorio.
C) Inflamación de etiología inmune y no relacionado directamente con los vasos: Este es el mecanismo que con mayor frecuencia se ve implicado en el desarrollo de las vasculitis, en donde la formación de complejos inmunes antígeno anticuerpo juega un papel fundamental.
Se han clasificado los mecanismos inmunes involucrados en cuatro tipos:
Tipo I (vasculitis alérgica o anafiláctica: incluye las vasculitis asociadas a estados atópicos, urticaria vasculitis y síndrome de Churg-Strauss. Se caracterizan por la producción de IgE, en respuesta a algún agente ambiental, que se unen a las células plasmáticas a través de su receptor Fc. En las exposiciones posteriores al agente ambiental, la IgE unida a las células plasmáticas induce la degranulación de los mastocitos, liberando mediadores que producen reacciones alérgicas. La fase vasculítica se caracteriza por infiltración angiocéntrica de los vasos por eosinófilos.
Tipo II (citotóxica o citolítica). Vasculitis mediadas por anticuerpos anticitoplasma de neutrófilos (ANCA) como la granulomatosis de Wegener, poliangeitis microscópica y síndrome de Churg-strauss. Los ANCA son capaces de activar los neutrófilos y las células endoteliales, así como inducir la apoptosis acelerada de los neutrófilos. Los anticuerpos anti-células endoteliales endoteliales (AECA) también pueden causar vascultis por daño directo o por activación del complemento, están involucrados en la enfermedad de Behçet y la enfermedad de Takayasu y tienen especificidad por diferentes regiones vasculares, afectando a vasos de pequeño tamaño en la enfermedad de Behçet y vasos de gran tamaño en la enfermedad de Takayasu.
Tipo III (mediada por inmunocomplejos): el depósito de inmunocomplejos da lugar a la activación del complemento y liberación de los componentes C3 y C5, que producen quimiotáxis de neutrófilos y liberación de enzimas proteolíticas que dañan la pared vascular. Es el grupo más amplio de vasculitis entre las que encontramos la vasculitis leucocitoclástica cutánea, el síndrome de Schonlein-Henoch y poliarteritis nodosa, entre otras.
Tipo IV (citotóxica) vasculitis mediada por linfocitos T: en este grupo se incluyen aquellas vasculitis granulomatosas que se caracterizan por la presencia en la pared de los vasos de infiltrados inducidos por linfocitos T, especialmente Th1, que serían responsables por medio de la producción de interferon-?, de la acumulación de macrófagos que fagocitarían las fibras elásticas. En este grupo de vasculitis se encuentra la arteritis de la temporal.
MANIFESTACIONES CLÍNICAS
La presencia de vasculitis se debe considerar en todos los pacientes que se presentan síntomas sistémicos y/o disfunción multiorgánica. Los síntomas son poco sensibles y específicos, e incluyen: fiebre, astenia, disnea, artralgias, dolor abdominal, hipertensión, insuficiencia renal, lesiones cutáneo mucosas, síntomas neurológicos, cardiopatía isquémica, afectación ocular, hepática, etc., retrasándose a menudo el diagnostico de vasculitis debido a que las manifestaciones clínicas pueden ser imitados por un gran número trastornos .
Las distintas vasculitis pueden presentar ciertos patrones de compromiso de órganos y sintomatología predominantes que orientará su diagnóstico diferencial:
Compromiso de pulmón y riñón (síndrome riñón-pulmón) en la granulomatosis de Wegener y en la poliangiitis microscópica. Sin embargo, algunos otros trastornos como la enfermedad por anticuerpos anti membrana basal glomerular (anti -MBG ), el lupus, etc. pueden causar una combinación idéntica de síntomas.
Compromiso piel y riñón (síndrome dérmico-renal) en la púrpura de Schönlein Henoch y en las crioglobulinemias.
Compromiso vía aérea superior en la granulomatosis de Wegener.
Compromiso pulmonar obstructivo, tipo asma bronquial, en la vasculitis de Churg-Strauss.
Compromiso intestinal, con dolor y hemorragia intestinal en la púrpura de Schönlein-Henoch
Ausencia de pulsos en extremidades en arteritis de Takayasu.
Cefalea, compromiso ocular y mandibular en la arteritis de la temporal.
Mononeuritis múltiple. Descartada la diabetes mellitus, la mononeuritis múltiple es altamente sugestiva de vasculitis (en particular la poliarteritis nodosa), ya que la neuropatía diabética es la única causa frecuente de este problema en los países desarrollados.
Púrpura palpable. La causa mas frecuente de púrpura palpable es la vasculitis leucocitoclástica cutánea por hipersensibilidad. Si la púrpura se acompaña de afectación de órganos sistémicos, es probable que el paciente padezca una vasculitis por IgA (purpua de Schönlein-Henoch), una poliangeítis microscópica, vasculitis asociada a ANCA, vasculitis secundaria, etc.
DIAGNÓSTICO
Anamnesis
Una anamnesis es importante para evaluar si al paciente utiliza fármacos (pueden producir vasculitis leucotoclstica por hipersensibilidad), tiene historia de hepatitis (la hepatitis C es responsable de la mayoría de los casos de crioglobulinemia y de algunos casos de poliarteritis microscópica), o ha sido diagnosticado de cualquier trastorno que se asocie a vasculitis secundaria: lupus eritematoso sistémico, artritis reumatoide, etc.
La mayor frecuencia de algunas vasculitis en ciertos grupos de edad y/o sexo pueden ser datos orientadores del diagnóstico. Así, la media de la edad de inicio es de 45 a 50 años en la granulomatosis de Wegener y en la poliarteritis nodosa, en comparación con los 17 y 26 años de edad edad media de presentación de la vasculitis por IgA (purpura de SH) y de la arteritis de Takayasu, o los 69 años de la arteritis de células gigantes. Además, estas dos últimas vasculitis se producen mas frecuentemente en las mujeres (86 y 75% respectivamente).
Exploración física
La exploración física ayuda a determinar la extensión de las lesiones vasculares, los órganos afectados y la presencia de una posible enfermedad primaria. Como se mencionó anteriormente, ciertos trastornos, como la mononeuritis múltiple y la púrpura palpable son altamente sugestivos de una vasculitis subyacente.
Exploraciones complementarias
El estudio inicial debe incluir determinaciones básicas de laboratorio, como hemograma, VS, PCR, creatinina, transaminasas, CPK, LDH, sistemático de orina, serología de hepatitis y radiografía de tórax y electrocardiograma. Otras pruebas de laboratorio adicionales, más específicas incluyen la determinación de:
ANA: Una prueba de anticuerpos antinucleares positiva sugiere la presencia de una enfermedad del tejido conectivo, particularmente lupus eritematoso sistémico.
Complemento C3 y C4: Niveles bajos de complemento pueden estar presentes en la crioglobulinemia y en el lupus.
En las personas con crioglobulinas positivas debe descartarse la infeccción por el virus de la hepatitis B y C.
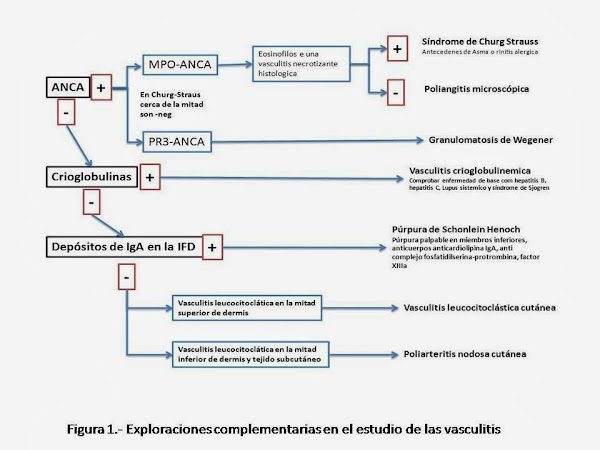
ANCA: Aunque no son diagnósticos, la presencia de ANCA dirigidos contra la proteasa 3 sugiere el diagnóstico de granulomatosis de Wegener, mientras que los ANCA dirigidos contra mieloperoxidasa son sugerentes de poliangeítis microscópica. También se asociada a ANCA positivos, principalmente pANCA, la poliangeitis granulomatosa eosinofílica ( S. de Churg-Strauss ), reacciones a fármacos y algunas infecciones y tumores. (Figura 1)
Electromiografía. El electromiografía es útil si hay síntomas y/o signos de sospecha de mononeuritis múltiple, polineuropatía o miopatía.
Biopsia de tejido. Idealmente se debe realizar biopsia del órgano comprometido para estudio histopatológico. La biopsia "a ciegas ", es menos probable que sea fructífera. Los órganos más frecuentemente biopsiados son la piel, arteria temporal, nervio sural, vía aérea superior (principalmente senos nasales y paranasales) y riñón. Menos frecuente, por la dificultad técnica que implica, el pulmón. Los hallazgos que se buscan para hacer el diagnóstico son la inflamación arterial, necrosis o granulomatosis. Siempre se complementa con inmunofluorescencia en busca de depósitos de IgA (Schönlein-Henoch), IgM e IgG (crioglobulinemias). Las vasculitis asociadas a ANCA son habitualmente pauci-inmunes y la inmunofluorescencia es negativa.
Arteriografía. Las arteriografías son útiles en la identificación y caracterización de una vasculitis de grandes y medianas arterias, como la poliarteritis nodosa, la arteritis de Takayasu y la arteritis de células gigantes con un síndrome del arco aórtico. En estos trastornos las alteraciones angiográficas no son patognomónica pero por lo general ayudan a establecer el diagnóstico cuando se combina con otros datos clínicos . Las angiografías de las arterias mesentéricas o renales en la poliarteritis nodosa puede mostrar aneurismas, oclusiones e irregularidades de la pared vascular. Por el contrario, la angiografía es improbable que sea útil en la evaluación de una vasculitis de pequeños vasos , como poliangeítis microscópico. Las técnicas de imagen más avanzadas, como la resonancia magnética, RM angiografía, angiografía por tomografía computarizada (TC) y doppler de troncos supraaórticos se utilizan cada vez con mas frecuencia para detectar lesiones de las arterias grandes, sin embargo, son menos sensible que la arteriografía convencional.
Otras pruebas complementarias se deben valorar según el caso, e incluyen el análisis del líquido cefalorraquídeo, hemocultivos, cultivos de distintos órganos, etc.
ACTITUD DIAGNÓSTICA ANTE UN PACIENTE CON SOSPECHA DE VASCULITIS EN ATENCIÓN PRIMARIA
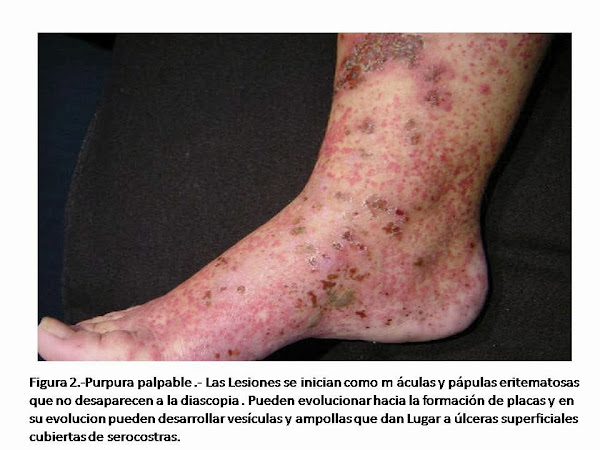
Con excepción de la arteritis de la temporal, la afectación de la piel y del tejido subcutáneo es la forma mas frecuente de presentación de las vasculitis en Atención Primaria, especialmente en forma de púrpura palpable (Figura 2). Sin embargo, la purpura palpable no es una manifestación específica de ningún tipo de vasculitis, pudiendo ser originada por una vasculitis infecciosa (vasculitis por neiseria), por una vasculitis mediada por inmunoclomplejos (por ejemplo la enfermedad del suero, las vasculitis por crioglobulinemia, o la púrpura de Henoch Schonlein), por una vasculitis asociada a ANCA ( granulomatosis de Wegener, poliangeritis microscópica), por una vasculitis alérgica (por una reacción medicamentosa), por una vasculitis asociada a una enfermedad reumatológica ( lupus eritematoso, arteritis reumatoide y síndrome de Sjogren) . De la misma manera, los nódulos cutáneos y subcutáneos inflamatorios (Figura 3) pueden ser originados por una gran variedad de vasculitis, incluyendo la poliarteritis nodosa, poliangitis microscópica, granulomatosis de Wegener, síndrome de Churg-Strauss.

Los pasos a realizar cuando existe la sospecha clínica de que estamos ante un paciente con una vasculitis cutánea deben estar dirigidos de la siguiente manera:
A) Confirmar el diagnóstico de vasculitis mediante la realización de una biopsia cutánea, ver el tipo de infiltrado, el tamaño del vaso afecto y realizar estudios de inmunofluorescencia directa para demostrar la presencia de inmunocomplejos.
B) Descartar la afectación de los órganos sistémicos. Los signos y síntomas que sugieren vasculitis en otros órganos incluyen:
Afectación muscular: Mialgias con elevación de enzimas musculares.
Afectación digestiva: Dolor abdominal con sangre oculta en feces o elevación de enzimas pancreáticos.
Afectación cardiaca: Angina con elevación de enzimas micocardicas.
Afectación renal: Hematuria con proteinuria, elevación de creatinina.
Afectación de nervios periféricos: Mononeuritis múltiple con defectos en la conducción nerviosa.
Afectación de sistema nervioso central: Disfunción cerebral o visual.
Test serológicos positivos: ANA, crioglobulinas, anticuerpos anti hepatitis B y C, ANCA, y niveles de complemento.
C) Estudiar la posible etiologia, intentado determinar si se trata de una vasculitis primaria o secundaria.
Descartar una causa infecciosa. Dado que el tratamiento de las vasculitis por infección directa de los vasos (vasculitis séptica) es completamente diferente de las vasculitis mediada inmunológicamente, esta causa debe ser descartada en el inicio de la valoración de un enfermo con vasculitis.
Si se determina que es una vasculitis mediada inmunológicamente debe determinarse si es de tipo III, mediada por inmunocomplejos, y deben buscarse el origen de estos inmunocomplejos, que puede ser exógeno (medicación, infección, proteínas) o exógeno (DNA, inmunoglobulinas, antígenos tumorales).
En la tabla 2.- se resumen las características clínicas y terapéuticas de las principales vasculitis primarias.
Tabla 2.- Diagnóstico y tratamineto de las vasculitis primarias
Arteritis de la Temporal
Clínica
Diagnóstico y tratamiento
Más frecuente en adultos mayores de 50 años y en mujeres que en hombres.
La cefalea suele ser el síntoma predominante. Se puede acompañar de hipersensibilidad del cuero cabelludo al peinarse o lavarse el pelo. En la exploración se pueden encontrar disminución del pulso de la arteria temporal y aumento de la sensibilidad.
Síntomas constitucionales frecuentes, con astenia, anorexia y perdida de peso.
Fiebre de grado variable en 15% de los casos.
Manifestaciones oftálmicas, hasta en 25-50% de los pacientes, secundarias a la oclusión de arterias orbitales u oftálmicas.
Claudicación mandibular, en hasta 70% de los pacientes. En forma ocasional se asocia a parestesias en la lengua, pérdida del gusto y dolor.
Otras menos frecuentes: hipoacusia, depresión, confusión, insuficiencia cardiaca
Hasta en el 50% de los pacientes se encuentran síntomas compatibles con una polimialgia reumática, que se manifiesta en su forma clásica con dolor en cintura pélvica y escapular. La polimialgia reumática puede presentarse aislada o acompañando a la microscópica. de la temporal.
Diagnóstico
Pruebas complemetarias: No hay marcador específico. La VS se encuentra elevada en el 90% de los pacientes.
Histopatología: La biopsia de la arteria temporal muestra característicamente un infiltrado inflamatorio granulomatoso, que compromete como panarteritis vasos de mediano y gran tamaño. El compromiso inflamatorio puede ser focal, lo que determina que la biopsia puede ser "negativa".
Tratamiento
El tratamiento con corticoides es de elección. En la arteritis de la temporal se debe iniciar tratamiento con prednisona 1 mg/kg/día, y posteriormente disminución gradual en un tiempo variable dependiendo de la respuesta clínica, habitualmente de 12-18 meses.
En la polimialgia reumática las dosis de de corticoides para el control de la enfermedad son menores, habitualmente iniciando con prednisona 20-30 mg/24 h, seguidos de una disminución gradual lenta, también en un período de 18-24 meses.
Arteritis de Takayasu
Clínica
Diagnóstico y tratamiento
Afecta predominantemente a mujeres jóvenes (razón mujer: hombre de 9:1.
Puede comprometer arco aórtico y sus ramas, pudiendo también comprometer aorta torácica, abdominal, arterias renales y arteria pulmonar, produciendo manifestaciones secundarias a la obstrucción de flujo sanguíneo de la arteria comprometida: claudicación de extremidades superiores (dificultad para levantar los brazos, peinarse, colgar ropa) y reducción o ausencia pulsos en la exploración física. Diferencia de al menos 10 mmHg en la presión arterial sistólica entre los brazos. Soplo sobre una o ambas arterias subclavias o aorta abdominal.
Pueden presentarse mareos, síncopes y alteraciones visuales.
Astenia, anorexia, pedida de peso y fiebre.
Artralgias y mialgias.
En etapas más tardías puede presentarse insuficiencia cardíaca y/o HTA de difícil manejo por estenosis de arterias renales.
Diagnóstico
Laboratorio: VS elevada en la mayoría de los pacientes.
Rx tórax: Puede presentarse un mediastino ensanchado a expensas del arco aórtico.
Arteriografía: estrechamiento u oclusión de la aorta, sus ramas primarias o grandes arterias de las extremidades proximales superiores o inferiores, no debidao arterioesclerosis, displasia fibromuscular, o por otras causas.
Histopatología: En general no se realiza biopsia por el tipo de vaso comprometido.
Tratamiento
En etapas precoces el tratamiento con corticoides (Prednisona 1 mg/kg/día) es fundamental. El tratamiento coadyuvante con bajas dosis de metotrexate (0,15-0,3 mg/kg/semanal vo) permite un buen control de la enfermedad y una reducción rápida de los corticoides
En etapas tardías frecuentemente es necesaria la cirugía, para reparar y excluir las arterias estenosadas.
Poliarteritis nodosa
Clínica
Diagnóstico y tratamiento
Aunque puede presentarse a cualquier edad, la media de la edad de presentación son los 50 años, afectando por igual a hombres y mujeres.
Mononeuritis múltiple: Neuropatía sensitiva y/o motora de predominio en MMII hasta en 70% de los pacientes, pudiendo ser la manifestación inicial de la enfermedad.
Lesiones cutáneas hasta en 60% de los pacientes: púrpura palpable, livedo reticularis, lesiones isquémicas en dedos, úlceras cutáneas, paniculitis y nódulos en el trayecto de los vasos.
Afectación renal con proteinuria de rango no nefrótico, cilindros hemáticos, hematuria franca, con HTA e insuficiencia renal.
Afectación aparato digestivo: dolor abdominal, alteración de transaminasas, pancreatitis, hemorragia digestiva y/o diarrea.
Compromiso testicular con dolor u orquitis.
Cardiomegalia en el 20% de los casos, cardiopatía isquémica e insuficiencia cardíaca.
Artralgias y artritis.
Síntomas constitucionales, pérdida de peso, fiebre y astenia.
Diagnóstico
Laboratorio: Es frecuente la anemia normocítica-normocrómica, trombocitosis y leucocitosis, elevación de la VS y PCR, elevación de la creatinina y alteraciones en el sedimento urinario.
Hasta en 50% de los casos hay antigenemia del virus de Hepatitis B (pudiendo ser portadores crónicos o presentar una hepatitis aguda por virus B).
Puede haber crioglobulinemia asociada. ANCA negativos y complemento normal
Arteriografia de la arteria renal o mesentérica: es útil para confirmar el diagnóstico y demuestra compromiso de arterias de mediano tamaño, alternando sitios de estenosis con dilataciones aneurismáticas y trombosis
Histopatología. La biopsia muscular a ciegas tiene una rentabilidad del 50-60%, y la dirigida mediante electromiograma del nervio sural del 90%.
En las fases agudas se evidencia edema de la íntima y necrosis fibrinoide de las arterias musculares con infiltrado de polimorfonucleares neutrófilos y ocasionalmente eosinófilos. En la fase crónica hay infiltrado mononuclear, engrosamiento de la íntima por fibrosis, destrucción de la lámina elástica, cicatrices fibrosis de la túnica media, estrechamiento de la luz trombosis y disección.
Tratamiento
Prednisona a dosis de 1 mg/kg/24h asociado o no a ciclofosfamida 2 mg/kg/24 h, dependiendo de la actividad y grado de extensión de la enfermedad.
Debe evaluarse presencia de hepatitis B para evaluar tratamiento con la combinación de agentes antivirales (interferón-alfa 2b o vidarabina), plasmaféresis y corticoides.
Enfermedad de Kawasaki
Clínica
Diagnóstico y tratamiento
Afecta preferentemente a niños menores de 2 años, afectando más frecuentemente a niños que a niñas.
Fiebre.
Conjuntivitis bulbar, no supurativa, bilateral.
Exantema cutáneo polimorfo.
Cambios en la mucosa oral: labios secos, rojos y fisurados, lengua aframbuesada, eritema orofaringeo.
Adenopatías cervicales mayores de 1,5 cm de diámetro.
Cambios en palmas de manos y plantas de pies: eritema y edema indurado. Posterior al décimo día de evolución se puede observar descamación de la punta de los dedos.
Artralgias y artritis en 20-30%
Uretritis y disuria
Compromiso neurológico: en forma de irritabilidad, convulsiones y meningitis aséptica
Compromiso gastrointestinal: dolor abdominal, vómitos y diarrea. Ictericia secundaria a hidrops vesicular. Puede haber ileo paralítico y alteración de pruebas hepática.s
Compromiso cardíaco que es el de mayor trascendencia- frecuente afectación arterias coronarias-: pericarditis, miocarditis, disfunción valvular e insuficiencia cardíaca.
Diagnóstico
Laboratorio: Frecuentemente VS y PCR elevadas, anemia normocítica-normocrómica y trombocitosis.
ECG: puede demostrar un intervalo PR y QT prolongado, cambios en segmentos ST y arritmias.
Rx tórax: puede demostrar cardiomegalia (secundaria a la miocarditis o pericarditis).
Ecocardiograma: puede demostrar cambios en las arterias coronarias como dilatación y aneurismas.
A los pacientes que presentan dolor torácico o evidencias de IAM se les debe realizar una coronariografía.
Histopatología: Afecta a arterias de mediano tamaño En la etapa inicial hay edema e infiltración de leucocitos y linfocitos. Posteriormente hay necrosis fibrinoide. Aneurismas con trombosis en vasos coronarios.
Tratamiento
El uso de gamaglobulina i. v. en forma precoz ha demostrado disminuir la frecuencia de dilataciones y aneurismas coronarios, especialmente el desarrollo de aneurismas mayores de 8 mm que son los que asocian a una mayor mortalidad. También debe tratarse con aspirina (3-5 mg/kg/día) como antiagregante plaquetario.
Vasculitis primaria del sistema nervioso central
Clínica
Diagnóstico y tratamiento
Enfermedad idiopática, recurrente, limitada al SNC y las meninges.
Trastorno infrecuente con predominio entre la cuarta y quinta décadas de la vida, aunque puede aparecer a cualquier edad.
Manifestaciones neurológicas heterogéneas:
a) Clínica neurológica multifocal que puede aparecer de forma secuencial y que remeda a la esclerosis múltiple, pero que además asocia signos y síntomas poco frecuentes en ésta, como las crisis comiciales o la cefalea
b) Forma de encefalopatía subaguda, con un síndrome confusional o una alteración de la consciencia.
c) Forma de lesión progresiva de características pseudotumoral.
Diagnóstico
Se caracteriza por una inflamación o necrosis de los vasos cerebrales de pequeño o mediano calibre.
Laboratorio: Exclusión de procesos infecciosos o inflamatorios sistémicos con los estudios apropiados de laboratorio.
Líquido cefalorraquídeo: pleocitosis y/o elevación de proteínas y que excluye infección o neoplasia.
RM: sugestivo de vasculitis del SNC y que excluya otras alternativas diagnósticas, seguido de una angiografía cerebral que demuestre estenosis segmentarias de vasos intracraneales.
Biopsia leptomeníngea y/o parenquimatosa cerebral que demuestra la existencia de inflamación vascular y que permite excluir otros diagnósticos.
Tratamiento
Los corticoides a altas dosis son el pilar clásico del tratamiento.
La adición de un agente citotóxico - ciclofosfamida, azatioprin o el metotrexato- ha mostrado mayor eficacia que los corticoides aislados, fundamentalmente en la prevención de recurrencias.
Poliarteritis microscópica
Clínica
Diagnóstico y tratamiento
Afecta por igual ambos sexos.
Es la causa más frecuente de síndrome riñón-pulmón
Sintomas constitucionales, fiebre, artralgias.
Compromiso cutáneo: púrpura, petequias, necrosis distal, úlceras.
Compromiso renal: glomerulonefritis en 90% de los pacientes, que puede acompañarse de insuficiencia renal.
Compromiso pulmonar con hemorragia alveolar.
Diagnóstico
Laboratorio: VS y PCR elevados. Hasta el 80% de los pacientes muestran ANCA +, principalmente pANCA, con ELISA + para el antígeno mieloperoxidasa (MPO)
Histopatología: Vasculitis necrotizante, sin formación de granulomas, similar que la la poliarteritis nodosa pero con compromiso de vaso pequeño
Tratamiento
El compromiso renal y pulmonar debe tratarse de forma agresiva con corticoides en dosis altas y ciclofosfamida iv. Cuando hay hemorragia alveolar, se debe añadir al tratamiento plasmaféresis.
Granulomatosis de Wegener
Clínica
Diagnóstico y tratamiento
Afecta a hombres y mujeres por igual, con edad media de presentación entre los 40-55 años.
Síntomas constitucionales, mialgias y artralgias
Compromiso cutáneo: nódulos subcutáneos, púrpura palpable, úlceras, vesículas o pápulas
Síntomas de vía aérea superior: síntomas nasales, sinusales, traqueales u óticos. Puede presentarse epistaxis, úlceras mucosas, perforación del septum nasal, deformaciones nasales (nariz en silla de montar), otitis media, parálisis nervio facial, estridor.
Afectación pulmonar, que puede ser un hallazgo a la radiografía en forma de nódulos o infiltrados, o ser sintomático con hemoptisis o insuficiencia respiratoria secundaria a hemorragia alveolar. Hasta 85% de los pacientes durante su evolución puede presentar este compromiso
Compromiso renal hasta en 75% de los casos, con sedimento de orina inflamatorio (hematuria dismórfica, cilindros hemáticos) e insuficiencia renal de grado variable.
Compromiso ocular: proptosis, diplopia, alteraciones de la mirada conjugada y pérdida visual.
Diagnóstico
Laboratorio: VS y PCR elevadas. Anemia normocitica normocromica de enfermedad crónica, trombocitosis. Hay presencia de cANCA hasta en 95% de los pacientes con enfermedad generalizada, con ELISA positivo para el antígeno del ANCAc, la proteinasa3 (PR3). El ANCA tiene valor diagnóstico en esta enfermedad, y también es un elemento útil para el seguimiento de la actividad de la enfermedad.
Histopatología: la biopsia renal demuestra en la mayoría de los casos una glomerulonefritis focal y segmentaria, que puede p
¿Vulnera este post tus derechos? Pincha aquí.
Creado: