La Leucemia Linfática Crónica (LLC) es una enfermedad caracterizada por la proliferación y acumulación de linfocitos de aspecto maduro en la sangre, la médula ósea, los ganglios linfáticos, el bazo, el hígado y otros órganos. Es la leucemia más común en los países occidentales, representando aproximadamente el 30 por ciento de todas ellas.
Es más común en hombres, con una relación hombre-mujer de aproximadamente 1,7:1. En Europa su tasa de incidencia anual se estima en 3-6 casos por 100.000 en los varones y 2-4 por 100.000 en las mujeres. La incidencia de LLC aumenta con la edad, afectando principalmente a personas mayores, con una edad media al diagnóstico de 70 años, sin embargo, no es inusual hacer el diagnóstico en individuos más jóvenes.
La LLC es una proliferación celular cuya naturaleza clonal se ha demostrado mediante técnicas de biología molecular, con el estudio de la reordenación de los genes que codifican las cadenas pesadas de las inmunoglobulinas.
En la actualidad se considera que LLC y el Linfoma Linfocítico de Células Pequeñas (LLCP) son la la misma enfermedad en diferente etapa evolutiva. Históricamente, el diagnóstico de LLCP se establecía mediante biopsia de los ganglios linfáticos en un paciente que se presentaba con linfadenopatías y sin linfocitosis periférica, mientras que el diagnostico de la LLC se establecía en pacientes con linfocitosis mediante el examen de la sangre periférica y la biopsia de la médula ósea. Actualmente, el diagnóstico de LLCP se establece en pacientes que muestran ganglios linfáticos con hallazgos coherente con LLC/LLCP pero con un recuento absoluto de linfocitos periféricos que no exceda 5.000 mm3 y sin evidencia de neutropenia, anemia, o trombocitopenia relacionada con la enfermedad.

MANIFESTACIONES CLÍNICAS
Las manifestaciones clínicas de la LLC se deben a la infiltración progresiva de la médula ósea, ganglios linfáticos y otros tejidos por linfocitos de pequeño tamaño, aspecto maduro y fenotipo B, así como a las alteraciones inmunológicas que acompañan a esta enfermedad. Sin embargo, en más de la mitad de los casos los pacientes están asintomáticos al inicio y su diagnóstico es un hallazgo incidental, en una analítica de rutina en la que aparece linfocitosis > 5.000/mm³ persistente más de 3 meses y que puede alcanzarse cifras muy superiores en el transcurso de la enfermedad. En otras ocasiones la LLC puede descubrirse a través del hallazgo (a veces por parte del propio paciente) de adenopatías superficiales. Con menor frecuencia, la enfermedad se diagnostica con motivo de complicaciones (infecciosas o hematológicas). A diferencia de lo que ocurre en los linfomas, la fiebre, la sudoración nocturna y la pérdida de peso no son frecuentes (10%). En la tabla I se resumen las principales manifestaciones de la LLC.
Tabla 1.- Principales manifestaciones de la LLC
· Linfocitosis > 5.000/mm³ persistente más de 3 meses
· Adenopatías: Presentes en el 50 a 90% de los pacientes.
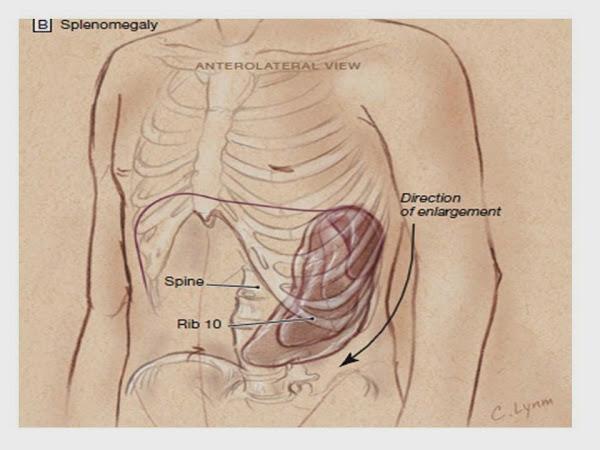
· Esplenomegalia: El bazo es el segundo órgano linfoide más frecuentemente afectado, palpable en el 25 al 55% de los casos y como es el caso de los ganglios linfáticos indoloro.
· Hepatomegalia: Presente en el 15 a 25% de los casos.
· Anemia: Generalmente, la anemia es de origen central y se debe a la infiltración de la médula ósea, lo que indica un estadio avanzado de la enfermedad. También, una anemia hemolítica autoinmune Coombs +, que provoca a veces cifras muy bajas de los niveles de hemoglobina puede poner de manifiesto la enfermedad o aparecer durante su evolución.
· Trombocitopenia: Lo más frecuente es que, como la anemia, se deba a la infiltración masiva de la médula ósea por linfocitos patológicos. A veces se debe a la destrucción periférica por anticuerpos, asociada o no a hiperesplenismo.
· Neutropenias absolutas: Más infrecuentes, se ven generalmente en los pacientes que se hayan sometido a una o varias quimioterapias. También indican la existencia de una enfermedad evolucionada. Las neutropenias de origen autoinmunitario son mucho más excepcionales.
· Lesiones en piel: La infiltración de la piel por células de la LLC puede ocurrir en cualquier momento a lo largo de su evolución, siendo la piel, en el momento del diagnóstico, el órgano no linfoide más comúnmente implicado, afectando las lesiones preferentemente a la cara, en forma de máculas, pápulas, placas, nódulos, úlceras o ampollas.
· Infecciones: En la enfermedad avanzada se observa una predisposición a las infecciones bacterianas, víricas y fúngicas como consecuencia de la hipogammaglobulinemia y la granulocitopenia.
· Trastornos inmunológicos: Frecuentemente la LLC se asocia a trastornos inmunológicos, como anemia hemolítica autoinmune, púrpura trombocitopénica de naturaleza inmune, y con una frecuencia mucho menor, aplasia selectiva eritropoyética o neutropenia secundarias a la producción de autoanticuerpos dirigidos contra las células progenitoras de esas líneas medulares. La prueba de Coombs resulta positiva en el 15 al 35 % de los casos, ya sea al diagnóstico o durante la evolución de la enfermedad. Es frecuente la hipogammaglobulinemia, siendo la inmunoglobulina que esta disminuida con mayor frecuencia la IgM, seguida de la IgG y de la IgA. En 5-10 % de los casos puede detectarse un componente monoclonal en el suero (frecuentemente IgM o IgG. También se han descrito alteraciones del complemento y de la actividad fagocítica.
DIAGNÓSTICO
La presencia de una linfocitosis superior a 5.000 mm³ ml y de marcadores de superficie característicos es necesaria y suficiente para establecer el diagnóstico de LLC.
El examen del frotis sanguíneo muestra el predominio de linfocitos pequeños, maduros, tipo B. En ausencia de enfermedad hay un 80% de linfocitos T y un 20% de linfocitos B, mientras que en la LLC esta relación se invierte (del 80 al 95% linfocitos B y del 5 al 20% de linfocitos T). Un porcentaje inferior al 10% de prolinfocitos y/o de linfocitos fragmentados no cuestiona el diagnóstico. Rara vez se observa neutropenia absoluta. La anemia y/o la trombocitopenia están presentes en el 15% de los casos.
El inmunofenotipo de las células linfocíticas sanguíneas se realiza mediante citometría de flujo de las células CD19 o CD20 con cada uno de los marcadores que figuran en la tabla 2. Permite establecer una puntuación inmunológica denominada puntuación de Matutes/Moreau o puntuación RMH (Royal Marsden Hospital), en función de la detección o no de estos diferentes marcadores de membrana. En la puntuación de Moreau, el CD79b sustituye al CD22.
Si la puntuación es igual o superior a 4, se establece el diagnóstico de LLC, y si es inferior a 3, debe descartarse. Si la puntuación es de 3, puede establecerse el diagnóstico de LLC si las células linfocíticas sanguíneas expresan las moléculas CD5, CD23 y CD43, si la expresión de CD20 es baja y si la búsqueda de la expresión de ciclina D1 es negativa.
Tabla 2 .- Puntuación inmunológica de Matutes/Moreau.
Valoración
1
0
CD5
+
-
CD23
+
-
Expresión Ig monotípica
Débil
Intensa
FMC7
-
+
Expresión de CD79b/CD22
Débil
Intensa
Existe frecuentemente hipogammaglobulinemia (hasta en el 60% de los casos), asociada a inmunoglobulina monoclonal (5% de los casos) de nivel sérico bajo.
La realización de biopsia de médula ósea no es necesaria para el diagnóstico, si bien sirve para completar el estudio. Confirmaría una linfocitosis medular superior al 30% y, se encontrarían todos los estadios, entre una infiltración nodular focal y una infiltración difusa, lo que indicaría una enfermedad más avanzada. De igual modo, no es imprescindible realizar una biopsia de una de las adenopatías afectas, salvo si se sospecha la transformación de la LLC a un síndrome de Richter
En el estudio citogenético realizado en la biopsia de medula ósea aparecen alteraciones cromosómicas, las más frecuentes son: la deleción del brazo largo del cromosoma 13 (13q-) en el 50% de los casos -variante típica-; la trisomia del cromosoma 12 (+12) entre el 10 y el 30% de los casos -variente atípica y proliferativa-; deleción en el cromosoma 11 entre el 10 y el 20% de los casos; deleción del cromosoma 17 en un 10% de los casos. También aparecen deleciones en el cromosoma 6 y translocaciones en el cromosoma 14.
Otras exploraciones complementarias
La realización de ecografía, TAC, RM, Gammagrafia, etc., según los casos sirven para apreciar el estado general del paciente en relación con la detección las adenopatías propias de la enfermedad y hepatoesplenomegalia.
Se debe estudiar comorbilidades, particularmente una insuficiencia renal; haptoglobina, bilirrubina, lactato deshidrogenasa (LDH); beta2-microglobulina, electroforesis de las proteínas séricas con inmunofijación; serología de las hepatitis B y C (riesgo de reactivación tras terapia inmunosupresora), La PET carece de utilidad a menos que se sospeche un síndrome de Richter.
En los ensayos clínicos, pueden ser útiles pruebas como el perfil mutacional del gen VH que codifica la región variable de las cadenas pesadas de las inmunoglobulinas y la expresión de ZAP-70, el cariotipo de los linfocitos sanguíneos, los marcadores de proliferación (timidina cinasa sérica o CD23 soluble, expresión de CD38) y la búsqueda mediante FISH de otras anomalías citogenéticas: deleción 13q y trisomía 12.
Los criterios diagnósticos de la LLC de la International Workshop on CLL, National Cancer Institute/Sponsored Working Group se resumen en la tabla 3.
Tabla 3.- Criterios diagnósticos de la LLC de la International Workshop on CLL, National Cancer Institute/Sponsored Working Group.
Linfocitosis mantenida superior a 5.000/ml
Morfología típica, con menos de un 10% de células de aspecto inmaduro
Fenotipo compatible con LLC (expresión de cadenas kappa o lambda; SmIg de poca intensidad, positividad para antígenos pan-B y el antígeno CD5)
Infiltración de la médula ósea superior al 30% y/o biopsia medular compatible con LLC.
El estudio de los marcadores celulares y de la médula ósea tiene especial importancia en los casos que cursan con linfocitosis inferiores a 5.000/ml.
Con la clasificación actual los pacientes con un recuento absoluto de linfocitos inferior a 5 x 10 9 /l, por lo general se diagnostican como una linfocitosis monoclonal B en lugar de LLC si no tienen adenopatías palpables, esplenomegalia, y/o hepatomegalia.
De acuerdo con la morfología de los linfocitos se distingue tres tipos de LLC: 1) LLC típica, con predominio de células pequeñas (más del 90%); 2) LLC atípica, en la que existe una proporción superior al 15% de linfocitos atípicos (centrocitos o células hendidas, linfoplasmocitos); 3) LLC con aumento de prolinfocitos (10-55%). La morfología atípica suele asociarse a alteraciones cromosómicas del tipo de la trisomía 12, y menos frecuentemente deleciones del brazo largo del cromosoma 6 (del6q).
CLASIFICACIÓN Y PRONOSTICO
La clasificación de la LLC en estadios clínicos resulta útil para definir el pronóstico y el tratamiento. En la actualidad se utilizan dos clasificaciones, la de Rai, que se basa principalmente en los cambios hematológicos, y la de Binet, que se fundamenta en la extensión de la enfermedad (Tabla 3).
Tabla 3.- Clasificación Rai y de Binet de la Leucemia Linfática Crónica
Clasificación de Rai
Estadio 0: Linfocitosis absoluta (>5.000/ml en SP) y >30% linfocitos en MO, sin adenopatía, hepatoesplenomegalia, anemia o trombocitopenia.
Estadio I: Estadio 0 con linfadenopatía y sin hepatoesplenomegalia, anemia o trombocitopenia.
Estadio II: Estadio I con hepatomegalia o esplenomegalia.
Estadio III: Estadio II y anemia (hemoglobina <11 g/dl)
Estadio IV: Estadio III con trombocitopenia (<100.000/ml)
Clasificación Binet ( Se utiliza más frecuentemente en Europa)
Estadio clínico A*: LLC en ausencia de anemia o trombocitopenia y menos de tres áreas de compromiso linfoide (estadios Rai 0, I, y II).
Estadio clínico B*: LLC en ausencia de anemia o trombocitopenia con tres o más áreas de implicación linfoide (estadios Rai I y II).
Estadio clínico C:LLC con anemia o trombocitopenia independientemente del número de áreas con aumento de volumen linfoide (estadios Rai III y IV).
* Las áreas linfoides incluyen la cervical, axilar, inguinal y el bazo.
La mediana de supervivencia de los pacientes con LLC es de aproximadamente 10 años, variando en función del estadio de 3 a 20 años.
Las principales complicaciones de la LLC son: a) Anemia hemolítica autoinmune; b) Infecciones bacterianas, víricas o fúngicas favorecidas por la hipogammaglobulinemia y el tratamiento; c) Transformación en linfoma de células grandes o en linfoma de Hodgkin (síndrome de Richter) -Entre 10 y 30 % de los pacientes con LLC sufren una transformación a un proceso linfoproliferativo más agresivo. La forma más frecuente de transformación es a una leucemia prolinfocitica, que ocurre en el 15-30 % de los casos y alrededor del 10 % de los pacientes desarrollan un linfoma agresivo (Síndrome de Richter). Sin embargo, es excepcional que la LLC acabe evolucionando hacia una leucemia aguda.- d) Cánceres secundarios o asociados: el estado de inmunodeficiencia predispone a la aparición de segundas neoplasias, siendo los tumores más frecuentes el carcinoma de piel, del tubo digestivo o de pulmón. También se han comunicado casos de LLC asociados a algunas hemopatías mieloides
TRATAMIENTO
Las indicaciones de tratamiento de la LLC dependerán esencialmente de tres criterios: la edad del paciente, el estado clínico y evolutivo de la enfermedad y la posible existencia de una deleción 17p.
Se encuentra actualmente bien establecido que no debe tratarse a los pacientes en estadio A de Binet. Podrían exceptuarse aquéllos que experimentan una evolución rápida del número de linfocitos circulantes (tiempo de duplicación inferior a un año, por encima de 50.000/mm3).
Tratamiento de primera línea
Pacientes sin deleción 17p
En los pacientes sin comorbilidad asociada, el tratamiento se basa en la asociación FCR (fludarabina, ciclofosfamida y rituximab).
En los pacientes con comorbilidad grave y en los pacientes de edad avanzada, el tratamiento persigue obtener la mejor calidad de vida posible, que depende de la calidad de la respuesta obtenida con el tratamiento. Los estudios aleatorizados demuestran la superioridad de la asociación FC (fludarabina y ciclofosfamida) en comparación con la fludarabina o el cloraminofeno en monoterapia y la falta de superioridad de la fludarabina en monoterapia en comparación con el cloraminofeno. Por ello se recomienda dar prioridad a la asociación FC o FCR ajustando las dosis según la función renal y disminuyendo el número de ciclos. Debido a la falta de respuesta duradera y de calidad, el cloraminofeno sólo se propone, con un efecto esencialmente sintomático, en los pacientes que no pueden tolerar los tratamientos mencionados anteriormente.
Pacientes con deleción 17p
Debido a la baja tasa de respuesta a la fludarabina y agentes alquilantes, se recomienda el tratamiento con alemtuzumab. Se utiliza solo o asociado a corticoides a dosis altas (dexametasona), en particular en los casos de masa tumoral ganglionar importante. En los pacientes jóvenes, debe considerarse rápidamente el aloinjerto de células madre hematopoyéticas.
Tratamiento de las recaídas
La elección del tratamiento depende de varios parámetros: edad del paciente; existencia de una deleción 17p, que debe buscarse mediante un nuevo examen citogenético; naturaleza del tratamiento o tratamientos previos, y duración de la última respuesta. Debe promoverse la inclusión en un ensayo clínico.
Primera recaída
En caso de recaída tardía, los tratamientos utilizados en primera instancia de tipo FCR se pueden usar de nuevo, salvo en caso de aparición de una deleción 17p. Si no se ha utilizado anteriormente el rituximab, se recomienda añadirlo.
Recaídas posteriores
Es preciso considerar la utilización de nuevos agentes, como la bendamustina. El aloinjerto de células madre hematopoyéticas está indicado en los pacientes más jóvenes en caso de recaída precoz después de FCR.
Formas refractarias
En caso de enfermedad refractaria (fracaso primario o remisión inferior a seis meses) o de progresión precoz (antes de 12 meses) tras tratamiento con F o FC o antes de 24 meses después del tratamiento con FCR, es razonable emplear alemtuzumab (±corticoides) o una asociación que incluya arabinósido de citosina a dosis altas y sales de platino. El aloinjerto de células madre hematopoyéticas está indicado en los pacientes jóvenes que presentan respuesta. En ausencia de donantes o si existe contraindicación para el aloinjerto, puede considerarse un autoinjerto.
Tratamiento de consolidación y mantenimiento
En estos momentos, ante la ausencia de estudios aleatorizados que permitan indicarlos, no deben considerarse los tratamientos de mantenimiento a menos que se realicen en el contexto de un ensayo terapéutico.
TRATAMIENTO DE LAS COMPLICACIONES
Citopenias autoinmunitarias
Anemia hemolítica autoinmunitaria (AHAI): Cuando aparece durante la evolución de una LLC, debe tratarse con corticoides (prednisona a dosis de 2 mg/kg/día) como tratamiento de primera línea. Si fracasa el tratamiento con corticoides y/o está indicado el tratamiento de la LLC, es posible un tratamiento con rituximab (R) y/o ciclofosfamida, R-mini-CHOP o alemtuzumab. La aparición de hemólisis contraindica definitivamente la utilización de la fludarabina en monoterapia. Es posible asociarla a ciclofosfamida y rituximab. La AHAI que se presenta durante un tratamiento que incluya fludarabina exige la interrupción de este fármaco.
Trombocitopenia periférica: En caso de trombocitopenia, es necesario realizar un mielograma. El tratamiento propuesto es el de una PTI si se trata de una trombocitopenia periférica (prednisona a dosis de 1 mg/kg y, en caso de fracaso o de trombocitopenia amenazante, perfusión de altas dosis de inmunoglobulinas). No es necesario tratar la LLC si es asintomática. Si fuese preciso tratarla, no debe utilizarse la fludarabina en monoterapia. La falta de respuesta a estos tratamientos puede conducir a una esplenectomía.
Eritroblastopenia: En caso de eritroblastopenia, hay que buscar una infección por parvovirus mediante la reacción en cadena de la polimerasa (PCR) en la médula ósea, ya que su existencia implica el tratamiento con inmunoglobulinas polivalentes. En los demás casos, se utiliza el tratamiento con corticoides (1-2 mg/kg) y, si no es eficaz, debe considerarse un tratamiento con ciclosporina.
Síndrome de Richter
Su tratamiento es el de un linfoma no Hodgkin de grado alto seguido de un autoinjerto o un aloinjerto.
Infección e hipogammaglobulinemia
Las sobreinfecciones, ya sean bacterianas o víricas, aparezcan después de la quimioterapia o sin que exista tratamiento, requieren un tratamiento antimicrobiano adecuado después de un estudio etiológico riguroso y rápido.
La perfusión regular de altas dosis de gammaglobulinas en pacientes que presentan hipogammaglobulinemia es discutible y su indicación sólo se reconoce en pacientes con infecciones recidivantes (sobre todo broncopulmonares). El contacto con un paciente con una infección vírica, como el virus de la varicela-zóster (VVZ), constituye una indicación puntual de gammaglobulinas.
PAPEL DEL MÉDICO DE FAMILIA
La gran mayoría de los pacientes son objeto de un seguimiento paralelo: en la consulta de los servicios de hematología y por el médico de familia, que tiene un papel importante.
Además de detectar la enfermedad cuando se produce uno de los episodios descritos anteriormente, es necesario un seguimiento clínico regular. Cualquier alteración del estado general, con o sin fiebre, debe llevar a buscar una complicación infecciosa o evolutiva de la enfermedad. La evolución de ésta es a menudo indolente y permite, la mayoría de las veces, proseguir con una vida normal con tratamientos que, por lo general, se toleran bien. En los pacientes sin tratamiento, se justifica la monitorización clínica dos veces al año con un hemograma (o incluso una vez al año si existe estabilidad). La vigilancia se refuerza si aparecen criterios de progresión clínica y biológica.
El seguimiento de los pacientes que han completado el tratamiento se realiza mediante la exploración física y el hemograma, con una periodicidad de tres a seis meses en función de su comorbilidad. El riesgo de infección persiste durante varios meses después del tratamiento, incluso si la respuesta ha sido muy buena. A menos que existan signos de alerta, no hay necesidad de repetir las exploraciones de diagnóstico por imagen ni el estudio de la enfermedad residual en ausencia de protocolo.
La prevención de las infecciones por VVZ y por Pneumocystis jirovecii se realiza, respectivamente, con valaciclovir y cotrimoxazol (o aerosoles de pentamidina o atovacuona en caso de alergia)hasta recuperar una concentración de linfocitos sanguíneos CD4+ superior a 200/mm3.
El tratamiento con alemtuzumab, debido al riesgo de reactivación de CMV, justifica la realización de una antigenemia o de una PCR-CMV, obligatoriamente en caso de fiebre y/o de síntomas sugestivos de enfermedad por CMV. La realización semanal de estas pruebas es cuestionable, ya que la confirmación de una reactivación asintomática no constituye una indicación sistemática para instaurar un tratamiento antivírico anticipatorio (ganciclovir o valganciclovir).
Los tratamientos que inducen una depleción notable y prolongada de los linfocitos CD4+ exponen a otros tipos de infecciones oportunistas (virus de Epstein-Barr, listeriosis, nocardiosis, infecciones por micobacterias, etc.).
BIBLIOGRAFÍA RECOMENDADA
Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981; 48:198.
Binet JL, Caligaris-Cappio F, Catovsky D, et al. Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 2006; 107:859.
Blum KA, Young D, Broering S, et al. Computed tomography scans do not improve the predictive power of 1996 national cancer institute sponsored working group chronic lymphocytic leukemia response criteria. J Clin Oncol 2007; 25:5624.
Boggs DR, Sofferman SA, Wintrobe MM, Cartwright GE. Factors influencing the duration of survival of patients with chronic lymphocytic leukemia. Am J Med 1966; 40:243.
Brachtl G, Piñón Hofbauer J, Greil R, Hartmann TN. The pathogenic relevance of the prognostic markers CD38 and CD49d in chronic lymphocytic leukemia. Ann Hematol 2014; 93:361.
Bulian P, Shanafelt TD, Fegan C, et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J Clin Oncol 2014; 32:897.
Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 2006; 24:437.
Call TG, Norman AD, Hanson CA, et al. Incidence of chronic lymphocytic leukemia and high-count monoclonal B-cell lymphocytosis using the 2008 guidelines. Cancer 2014; 120:2000.
Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists Collaborative Group. J Natl Cancer Inst 1999; 91:861.
Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2002; 100:4609.
Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996; 87:4990.
Chevallier P, Penther D, Avet-Loiseau H, et al. CD38 expression and secondary 17p deletion are important prognostic factors in chronic lymphocytic leukaemia. Br J Haematol 2002; 116:142.
Chronic lymphocytic leukemia: recommendations for diagnosis, staging, and response criteria. International Workshop on Chronic Lymphocytic Leukemia. Ann Intern Med 1989; 110:236.
Claus R, Lucas DM, Ruppert AS, et al. Validation of ZAP-70 methylation and its relative significance in predicting outcome in chronic lymphocytic leukemia. Blood 2014; 124:42.
Cui B, Chen L, Zhang S, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014; 124:546.
Damle RN, Ghiotto F, Valetto A, et al. B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood 2002; 99:4087.
Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94:1840.
Deaglio S, Vaisitti T, Aydin S, et al. In-tandem insight from basic science combined with clinical research: CD38 as both marker and key component of the pathogenetic network underlying chronic lymphocytic leukemia. Blood 2006; 108:1135.
Del Giudice I, Chiaretti S, Tavolaro S, et al. Spontaneous regression of chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood 2009; 114:638.
Del Poeta G, Maurillo L, Venditti A, et al. Clinical significance of CD38 expression in chronic lymphocytic leukemia. Blood 2001; 98:2633.
Del Principe MI, Del Poeta G, Buccisano F, et al. Clinical significance of ZAP-70 protein expression in B-cell chronic lymphocytic leukemia. Blood 2006; 108:853.
Delgado J, Pratt G, Phillips N, et al. Beta2-microglobulin is a better predictor of treatment-free survival in patients with chronic lymphocytic leukaemia if adjusted according to glomerular filtration rate. Br J Haematol 2009; 145:801.
Dicker F, Schnittger S, Haferlach T, et al. Immunostimulatory oligonucleotide-induced metaphase cytogenetics detect chromosomal aberrations in 80% of CLL patients: A study of 132 CLL cases with correlation to FISH, IgVH status, and CD38 expression. Blood 2006; 108:3152.
Dickinson JD, Smith LM, Sanger WG, et al. Unique gene expression and clinical characteristics are associated with the 11q23 deletion in chronic lymphocytic leukaemia. Br J Haematol 2005; 128:460.
Dreger P, Stilgenbauer S, Benner A, et al. The prognostic impact of autologous stem cell transplantation in patients with chronic lymphocytic leukemia: a risk-matched analysis based on the VH gene mutational status. Blood 2004; 103:2850.
Fong D, Kaiser A, Spizzo G, et al. Hodgkins disease variant of Richters syndrome in chronic lymphocytic leukaemia patients previously treated with fludarabine. Br J Haematol 2005; 129:199.
French Cooperative Group on Chronic Lymphocytic Leukaemia. Natural history of stage A chronic lymphocytic leukaemia untreated patients. Br J Haematol 1990; 76:45.
Friedman DR, Weinberg JB, Barry WT, et al. A genomic approach to improve prognosis and predict therapeutic response in chronic lymphocytic leukemia. Clin Cancer Res 2009; 15:6947.
Galton DA. The pathogenesis of chronic lymphocytic leukemia. Can Med Assoc J 1966; 94:1005.
Gentile M, Mauro FR, Calabrese E, et al. The prognostic value of CD38 expression in chronic lymphocytic leukaemia patients studied prospectively at diagnosis: a single institute experience. Br J Haematol 2005; 130:549.
Gentile M, Mauro FR, Rossi D, et al. Italian external and multicentric validation of the MD Anderson Cancer Center nomogram and prognostic index for chronic lymphocytic leukaemia patients: analysis of 1502 cases. Br J Haematol 2014; 167:224.
Giles FJ, OBrien SM, Keating MJ. Chronic lymphocytic leukemia in (Richters) transformation. Semin Oncol 1998; 25:117.
Grabowski P, Hultdin M, Karlsson K, et al. Telomere length as a prognostic parameter in chronic lymphocytic leukemia with special reference to VH gene mutation status. Blood 2005; 105:4807.
Grever MR, Lucas DM, Dewald GW, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 2007; 25:799.
Gribben JG. How I treat CLL up front. Blood 2010; 115:187.
Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446.
Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94:1848.
Hamblin TJ, Davis ZA, Oscier DG. Determination of how many immunoglobulin variable region heavy chain mutations are allowable in unmutated chronic lymphocytic leukaemia - long-term follow up of patients with different percentages of mutations. Br J Haematol 2008; 140:320.
Ibrahim S, Keating M, Do KA, et al. CD38 expression as an important prognostic factor in B-cell chronic lymphocytic leukemia. Blood 2001; 98:181.
International CLL-IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol 2016; 17:779.
Kanzler H, Küppers R, Helmes S, et al. Hodgkin and Reed-Sternberg-like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell-derived clones: potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkins disease. Blood 2000; 95:1023.
Keating MJ, Scouros M, Murphy S, et al. Multiple agent chemotherapy (POACH) in previously treated and untreated patients with chronic lymphocytic leukemia. Leukemia 1988; 2:157.
Keller JW, Knospe WH, Raney M, et al. Treatment of chronic lymphocytic leukemia using chlorambucil and prednisone with or without cycle-active consolidation chemotherapy. A Southeastern Cancer Study Group Trial. Cancer 1986; 58:1185.
Khouri IF, Saliba RM, Admirand J, et al. Graft-versus-leukaemia effect after non-myeloablative haematopoietic transplantation can overcome the unfavourable expression of ZAP-70 in refractory chronic lymphocytic leukaemia. Br J Haematol 2007; 137:355.
Lipshutz MD, Mir R, Rai KR, Sawitsky A. Bone marrow biopsy and clinical staging in chronic lymphocytic leukemia. Cancer 1980; 46:1422.
Maddocks-Christianson K, Slager SL, Zent CS, et al. Risk factors for development of a second lymphoid malignancy in patients with chronic lymphocytic leukaemia. Br J Haematol 2007; 139:398.
Mainou-Fowler T, Dignum H, Taylor PR, et al. Quantification improves the prognostic value of CD38 expression in B-cell chronic lymphocytic leukaemia. Br J Haematol 2002; 118:755.
Majid A, Lin TT, Best G, et al. CD49d is an independent prognostic marker that is associated with CXCR4 expression in CLL. Leuk Res 2011; 35:750.
Manshouri T, Do KA, Wang X, et al. Circulating CD20 is detectable in the plasma of patients with chronic lymphocytic leukemia and is of prognostic significance. Blood 2003; 101:2507.
Maurer MJ, Cerhan JR, Katzmann JA, et al. Monoclonal and polyclonal serum free light chains and clinical outcome in chronic lymphocytic leukemia. Blood 2011; 118:2821.
Melo JV, Catovsky D, Galton DA. The relationship between chronic lymphocytic leukaemia and prolymphocytic leukaemia. II. Patterns of evolution of prolymphocytoid transformation. Br J Haematol 1986; 64:77.
Michallet M, Archimbaud E, Bandini G, et al. HLA-identical sibling bone marrow transplantation in younger patients with chronic lymphocytic leukemia. European Group for Blood and Marrow Transplantation and the International Bone Marrow Transplant Registry. Ann Intern Med 1996; 124:311.
Moreno C, Hodgson K, Ferrer G, et al. Autoimmune cytopenia in chronic lymphocytic leukemia: prevalence, clinical associations, and prognostic significance. Blood 2010; 116:4771.
Morrison WH, Hoppe RT, Weiss LM, et al. Small lymphocytic lymphoma. J Clin Oncol 1989; 7:598.
Muntañola A, Bosch F, Arguis P, et al. Abdominal computed tomography predicts progression in patients with Rai stage 0 chronic lymphocytic leukemia. J Clin Oncol 2007; 25:1576.
Nabhan C, Coutré S, Hillmen P. Minimal residual disease in chronic lymphocytic leukaemia: is it ready for primetime? Br J Haematol 2007; 136:379.
Oscier DG, Gardiner AC, Mould SJ, et al. Multivariate analysis of prognostic factors in CLL: clinical stage, IGVH gene mutational status, and loss or mutation of the p53 gene are independent prognostic factors. Blood 2002; 100:1177.
Parikh SA, Strati P, Tsang M, et al. Should IGHV status and FISH testing be performed in all CLL patients at diagnosis? A systematic review and meta-analysis. Blood 2016; 127:1752.
Pepper C, Majid A, Lin TT, et al. Defining the prognosis of early stage chronic lymphocytic leukaemia patients. Br J Haematol 2012; 156:499.
Pflug N, Bahlo J, Shanafelt TD, et al. Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood 2014; 124:49.
Queirós AC, Villamor N, Clot G, et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2015; 29:598.
Rai KR, Chiorazzi N. Determining the clinical course and outcome in chronic lymphocytic leukemia. N Engl J Med 2003; 348:1797.
Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46:219.
Rai KR. A critical analysis of staging in CLL. In: Chronic Lymphocytic Leukemia: Recent Progress and future Direction. 1987 UCLA Symposia on Molecular and Cellular Biology, New Series, Vol. 59. Gale RP, Rai KR (Eds). Alan R Liss, New York 1987. p.253.
Rassenti LZ, Jain S, Keating MJ, et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood 2008; 112:1923.
Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001; 194:1639.
Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013; 121:1403.
Rossi D, Terzi-di-Bergamo L, De Paoli L, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood 2015; 126:1921.
Shanafelt TD, Byrd JC, Call TG, et al. Narrative review: initial management of newly diagnosed, early-stage chronic lymphocytic leukemia. Ann Intern Med 2006; 145:435.
Shanafelt TD, Drake MT, Maurer MJ, et al. Vitamin D insufficiency and prognosis in chronic lymphocytic leukemia. Blood 2011; 117:1492.
Shanafelt TD, Geyer SM, Kay NE. Prognosis at diagnosis: integrating molecular biologic insights into clinical practice for patients with CLL. Blood 2004; 103:1202.
Solh M, Rai KR, Peterson BL, et al. The impact of initial fludarabine therapy on transformation to Richter syndrome or prolymphocytic leukemia in patients with chronic lymphocytic leukemia: analysis of an intergroup trial (CALGB 9011). Leuk Lymphoma 2013; 54:252.
Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood 2014; 123:3247.
Tinhofer I, Rubenzer G, Holler C, et al. Expression levels of CD38 in T cells predict course of disease in male patients with B-chronic lymphocytic leukemia. Blood 2006; 108:2950.
Tsimberidou AM, Wen S, OBrien S, et al. Assessment of chronic lymphocytic leukemia and small lymphocytic lymphoma by absolute lymphocyte counts in 2,126 patients: 20 years of experience at the University of Texas M.D. Anderson Cancer Center. J Clin Oncol 2007; 25:4648.
Weisser M, Yeh RF, Duchateau-Nguyen G, et al. PTK2 expression and immunochemotherapy outcome in chronic lymphocytic leukemia. Blood 2014; 124:420.
Wierda WG, OBrien S, Wang X, et al. Characteristics associated with important clinical end points in patients with chronic lymphocytic leukemia at initial treatment. J Clin Oncol 2009; 27:1637.
Wierda WG, OBrien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011; 29:4088.
Zent CS, Ding W, Schwager SM, et al. The prognostic significance of cytopenia in chronic lymphocytic leukaemia/small lymphocytic lymphoma. Br J Haematol 2008; 141:615.
¿Vulnera este post tus derechos? Pincha aquí.
Creado: