La anemia no es mas que la disminución de glóbulos rojos y de hemoglobina, disminuyendo la capacidad de oxigeno; cuando las cifras de hemoglobina bajas a 7 mg/dl se aprecian una serie de manifestaciones clínicas como la palidez. La anemia no es una enfermedad sino consecuencias de procesos patológicos. En nuestro post te mencionamos los tipos de anemias y de que manera repercuten en el organismo.
Tipos de anemias
1-.Anemia hipoplasica congénita
La anemia hipoplásica congénita (anemia de Blackfan-Diamond) es una rara enfermedad unos de los tipos de anemia, que suele hacerse sintomática en la infancia temprana, a menudo con palidez en el período neonatal, aunque en ocasiones se manifiesta en etapas más avanzadas de la infancia.
Más del 90% de los casos se manifiestan en el primer año de vida, siendo la edad media al diagnóstico de 3 meses. Los rasgos hematológicos más característicos son anemia, generalmente macrocítica, reticulocitopenia y deficiencia o ausencia de precursores eritroides en una médula ósea que, por lo demás, muestra una celularidad normal.
Síntomas
Aunque durante la vida fetal la hematopoyesis suele ser adecuada, algunos lactantes afectados muestran palidez al nacer o durante los primeros días de vida; y en raros casos aparece hidropesía fetal. Hacia los 2 a 6 meses de vida se desarrolla una intensa anemia, que en ocasiones puede aparecer más tarde.
Más del 50% de los niños afectados tienen otras anomalías congénitas tales como baja talla, dismorfismo craneofacial (nariz aplastada, hipertelorismo, labio superior grueso) o defectos de las extremidades superiores (debilidad del pulso radial, aplanamiento de la eminencia tenar), entre ellos pulgares trifalángicos. Las anomalías son variadas, sin manifestarse un patrón específico en la mayoría de los afectados
-Pruebas de laboratorio
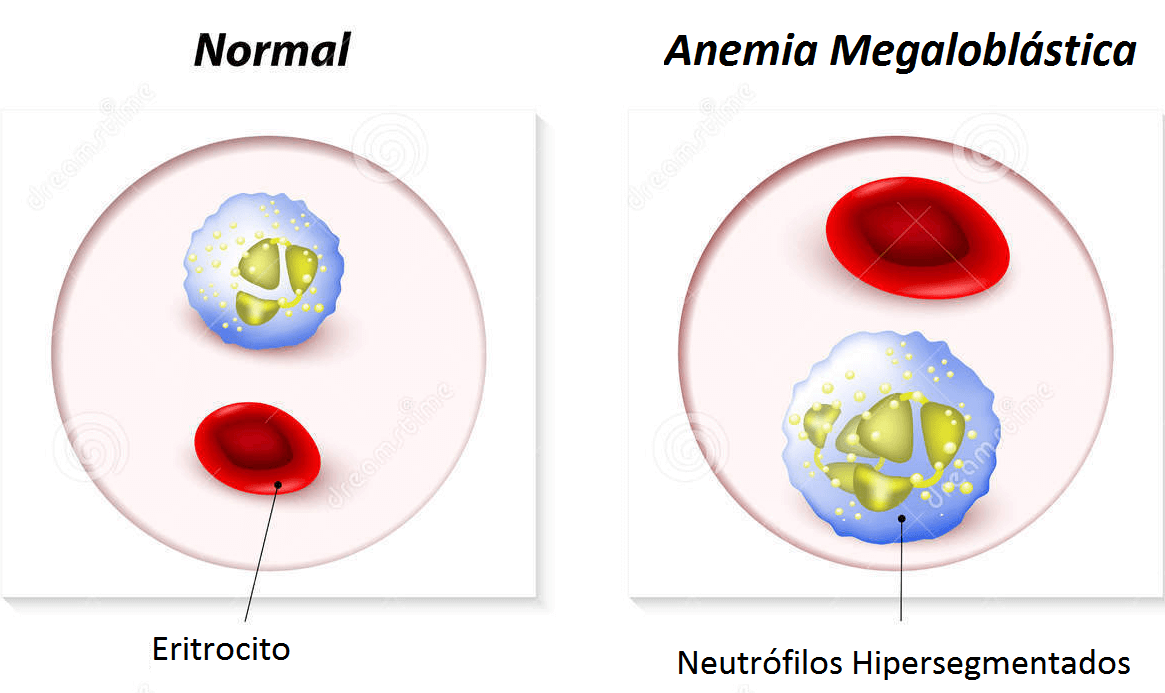
Los eritrocitos suelen ser macrocíticos para la edad del paciente, aunque no se observan la hipersegmentación de los neutrófilos ni otras alteraciones de la sangre periférica características de las anemias megaloblásticas. Las concentraciones de ácido fólico y de vitamina B12 son normales.
El análisis químico de los eritrocitos revela un patrón enzimático similar al de la población eritrocitaria «fetal», y además hay aumento de la hemoglobina fetal (Hb F) y mayor expresión de antígeno «i». La actividad de la adenosina desaminasa (ADA) hemática está aumentada en la mayoría de los pacientes con este cuadro, un hallazgo que ayuda a distinguir la aplasia eritrocitaria
Tratamiento
La administración de corticosteroides es beneficiosa en 3/4 partes de los pacientes que responden inicialmente. El mecanismo delos tipos de anemias este efecto se desconoce. Como prueba inicial, se administra prednisona en dosis de 2 mg/kg/24 horas dividido en tres dosis.
Entre 1 y 3 semanas después de la instauración del tratamiento, se observa un incremento de los precursores eritroides en la médula ósea, seguido de una reticulocitosis periférica. La hemoglobina puede alcanzar cifras normales en 4 a 6 semanas, aunque la tasa de respuesta es muy variable.
Una vez que se ha comprobado que la concentración de hemoglobina está aumentando, puede reducirse la dosis de corticosteroides de manera gradual, reduciendo cada una de la toma para acabar dejando la menor dosis efectiva diaria, que se administra en una sola toma.
Pronostico
Es probable que la supervivencia media sea >40 años, si bien no se dispone de datos definitivos. El registro de los pacientes con anemia de Blackfan-Diamond (DBAR) está acumulando información que podrá dar respuesta a datos sobre el tratamiento o la supervivencia
La evolución es mejor en los pacientes de los tipos de anemias que responden a los corticosteroides, y alrededor de la 1/2 de los pacientes tienen respuestas prolongadas. En los demás, la supervivencia depende de las transfusiones. Algunos niños de los dos grupos desarrollan remisiones espontáneas (20%) que, en la mayoría de los casos, suceden en el primer decenio de la vida. En los niños que reciben transfusiones regulares, el hierro orgánico total aumenta y aparece hemosiderosis.

La anemia de Blackfan-Diamond puede ser un síndrome premaligno y un pequeño porcentaje de pacientes (<5%) evolucionan hacia una leucemia aguda (en general mieloide) o una mielodisplasia como unos de los tipos de anemias mas raras. También se han descrito tumores sólidos malignos, en especial osteosarcomas. Otras causas importantes de muerte son las complicaciones asociadas al trasplante de células precursoras, al tratamiento corticoideo (infecciones oportunistas) y a la sobrecarga de hierro.
2-.Eritrobalastopenia
La aplasia eritrocitaria adquirida infantil más frecuente, la eritroblastopenia transitoria infantil (ETI), posee una incidencia superior a la de la anemia hipoplásica congénita.
Este síndrome de anemia hipoplásica transitoria grave afecta sobre todo a niños previamente sanos de 6 meses a 3 años de edad, siendo la mayoría mayores de 12 meses otros de los tipos de anemias. Sólo el 10% son > de 3 años. La causa de esta disminución de la producción de eritrocitos es una supresión inmunitaria transitoria de la eritropoyesis.
A menudo aparece tras una enfermedad viral, aunque no se ha identificado ningún virus específico. Las infecciones por parvovirus B19 (v. cap. 248), que pueden producir hipoplasia en niños con anemia hemolítica crónica no son las responsables de la ETI.
3-.Aplasia eritrocitaria asociada hemolisis
Causa viral mejor conocida de aplasia eritrocitaria en los fetos, los pacientes inmunodeprimidos o en los pacientes con hemólisis crónica es el parvovirus B19, agente productor de la quinta enfermedad o eritema infeccioso.

En el estudio con microscopia óptica de la médula ósea pueden verse las inclusiones nucleares características en los eritroblastos y en pronormoblastos gigantes. Como la infección por este virus suele ser transitoria y la recuperación tiene lugar en <2 semanas, los niños por lo demás normales, en los que la esperanza de vida de los hematíes es de 100 a 120 días, no sufren anemia o no es detectable.
Sin embargo, en los pacientes con hemólisis dentro de los tipos de anemias como la causada por una esferocitosis hereditaria o una drepanocitosis, en los que la esperanza de vida de los hematíes es mucho menor, una breve interrupción de la eritropoyesis debida a la infección por el parvovirus puede dar lugar a una anemia severa que se traduce en las llamadas crisis aplásicas características de estas enfermedades.
4-.Aplasia eritrocitaria
En raras ocasiones, la infección por parvovirus persiste en pacientes que no pueden desarrollar una respuesta adecuada de anticuerpos frente al virus, como en niños con inmunodeficiencias congénitas, en los que reciben tratamiento con agentes inmunosupresores o en los que padecen SIDA.
La aplasia eritrocitaria pura resultante puede ser grave dentro de los tipos de anemias y a menudo se cree que el niño afectado sufre una ETI dentro de los tipos de anemias. Esta modalidad de aplasia eritrocitaria difiere de la ETI en la ausencia de recuperación espontánea y en la necesidad de administrar más de una transfusión.
El diagnóstico de infección por parvovirus se hace mediante la detección de las partículas virales (por reacción en cadena de la polimerasa), ya que las respuestas serológicas habituales están disminuidas en los pacientes con inmunodeficiencia. El tratamiento de la infección viral en estos casos crónicos puede hacerse con inmunoglobulina intravenosa (IGIV) en dosis altas, que contiene anticuerpos neutralizantes del parvovirus
5-.Aplasia eritrocitaria con aborto espontáneo e hidropesía fetal.
Las infecciones intrauterinas por parvovirus y la consiguiente destrucción de los precursores eritroides causan distintas manifestaciones clínicas, con aumento de la frecuencia de abortos espontáneos en el primer y segundo trimestres y de recién nacidos con hidropesía fetal y viremia.
La presencia de infección congénita persistente por parvovirus se detecta mediante la reacción en cadena de la polimerasa o estudiando el ADN de la médula ósea y/o de la sangre periférica, ya que la tolerancia inmunitaria al virus puede evitar el desarrollo normal de anticuerpos específicos.
6-.Otras aplasias eritrocitarias infantiles.
Las aplasias eritrocitarias de tipo adulto suelen ser crónicas y mediadas por anticuerpos y a menudo se asocian a trastornos tales como la leucemia linfoide crónica, linfomas, timomas, trastornos linfoproliferativos y lupus eritematoso sistémico. Este tipo de aplasia eritrocitaria crónica producida por anticuerpos es muy rara en la infancia.
El alemtuzumab (anticuerpo anti CD52 humanizado) se ha utilizado para tratar la aplasia eritrocitaria pura de inicio en la edad adulta cuando el tratamiento con corticosteroides y otros agentes inmunosupresores ha fracasado.

Algunos fármacos, como el cloranfenicol, también pueden inhibir la eritropoyesis de forma dependiente de la dosis y sus efectos en la médula ósea se manifiestan por reticulocitopenia, hipoplasia eritroide y pronormoblastos vacuolados; estas manifestaciones difieren de las de la anemia aplásica idiosincrásica severa, que sucede con muy poca frecuencia en pacientes en tratamiento con cloranfenicol.
Te interesara leer: Anemia en Niños : Fisiológico o Patológico
.