Las anemias hemolíticas inmunitaria más importante en pediatría es la enfermedad hemolítica del recién nacido (eritroblastosis fetal), causada por el paso transplacentario de anticuerpos maternos activos frente a los hematíes del feto, es decir, una anemia hemolítica isoinmunitaria. Existen otras diversas anemias hemolíticas que son de origen autoinmunitario y que pueden ser idiopáticas o relacionadas con diversas infecciones (virus Epstein-Barr, rara vez VIH, citomegalovirus y micoplasma), con enfermedades de origen inmunitario (lupus eritematoso diseminado [LED], artritis reumatoide), con inmunodeficiencia (agammaglobulinemia, trastornos linfoproliferativos autoinmunitarios y disgammaglobulinemias), con neoplasias (linfoma, leucemia y enfermedad de Hodgkin) y con fármacos (metildopa, L-dopa).
Otros medicamentos (penicilinas, cefalosporinas) producen una hemólisis inmunitaria que no es de tipo autoinmunitario, sino debida a anticuerpos «dependientes del fármaco» que, aunque a veces lo son, no suelen ser «específicos» para los antígenos de la membrana eritrocitaria.
En el patrón habitual de deficiencia de G6PD, los síntomas se desarrollan entre 24 y 48 horas después de ingerir una sustancia con propiedades oxidantes. Los fármacos que tienen estas propiedades son la aspirina, las sulfamidas y los antipalúdicos tales como la primaquina (tabla 463-1). En algunos pacientes, la ingestión de habas, un producto alimenticio importante en la región mediterránea, también puede producir un síndrome hemolítico agudo llamado fabismo, provocado por los oxidantes derivados de 2 componentes glucosídicos, la vicina y la convicina, que se hidrolizan a divicina y a isouramilo y que, en último término, producen peróxido de hidrógeno y otros productos reactivos del oxígeno.
¿A qué se debe la anemia?
En las anemias hemolíticas autoinmunitarias, los anticuerpos anormales se dirigen contra los eritrocitos, pero los mecanismos patogénicos son dudosos. El autoanticuerpo podría deberse a una respuesta inmunitaria incorrecta frente a un antígeno eritrocitario o a otro epítopo antigénico similar a un antígeno eritrocitario, fenómeno conocido como mimetismo molecular. También es posible que un agente infeccioso altere la membrana del hematíe y la convierta en antigénica o «extraña » para el huésped.

Los anticuerpos suelen reaccionar contra epítopos (antígenos) que son «públicos» o comunes a todos los eritrocitos humanos como, por ejemplo, las proteínas Rh. En la mayoría de los casos de hemólisis por anticuerpos calientes no puede encontrarse ninguna causa subyacente y se habla de enfermedades primarias o idiopáticas . Si la hemólisis autoinmunitaria se asocia a una enfermedad subyacente como, por ejemplo, un trastorno linfoproliferativo, un LED o una inmunodeficiencia, recibirá el calificativo de secundaria. Los fármacos podrían estar implicados hasta en el 20% de los casos de hemólisis inmunitaria. Los medicamentos (penicilinas o a veces cefalosporinas) que causan hemólisis a través de un mecanismo de «hapteno».
Síntomas
Las anemias hemolíticas autoinmunitarias pueden manifestarse con 2 patrones clínicos generales. El primero, una forma transitoria aguda que dura de 3 a 6 meses y que afecta sobre todo a niños de 2 a 12 años de edad; constituye del 70 al 80% de los casos. A menudo va precedido de una infección, en general respiratoria.
El comienzo puede ser agudo, con postración, palidez, ictericia, pirexia y hemoglobinuria, o más gradual, con un cuadro en el que predominan la fatiga y la palidez.
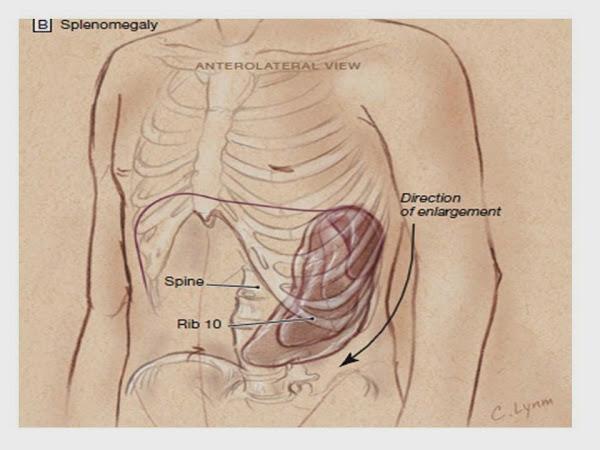
El bazo suele estar aumentado de tamaño y es el lugar fundamental donde se destruyen los eritrocitos recubiertos por la inmunoglobulina G (IgG). Las enfermedades sistémicas subyacentes son poco frecuentes. Otras características de la forma aguda son la respuesta constante a los glucocorticoides, la reducida tasa de mortalidad y la recuperación total. El otro patrón clínico conlleva un curso crónico y prolongado y es mucho más común en lactantes y niños mayores de 12 años. La hemólisis puede prolongarse durante muchos meses o años. Son frecuentes las alteraciones de los demás elementos formes sanguíneos y la respuesta a los glucocorticoides es variable e inconstante. La mortalidad es aproximadamente del 10% y la muerte a menudo puede atribuirse a la enfermedad sistémica subyacente.

Pruebas de laboratorio
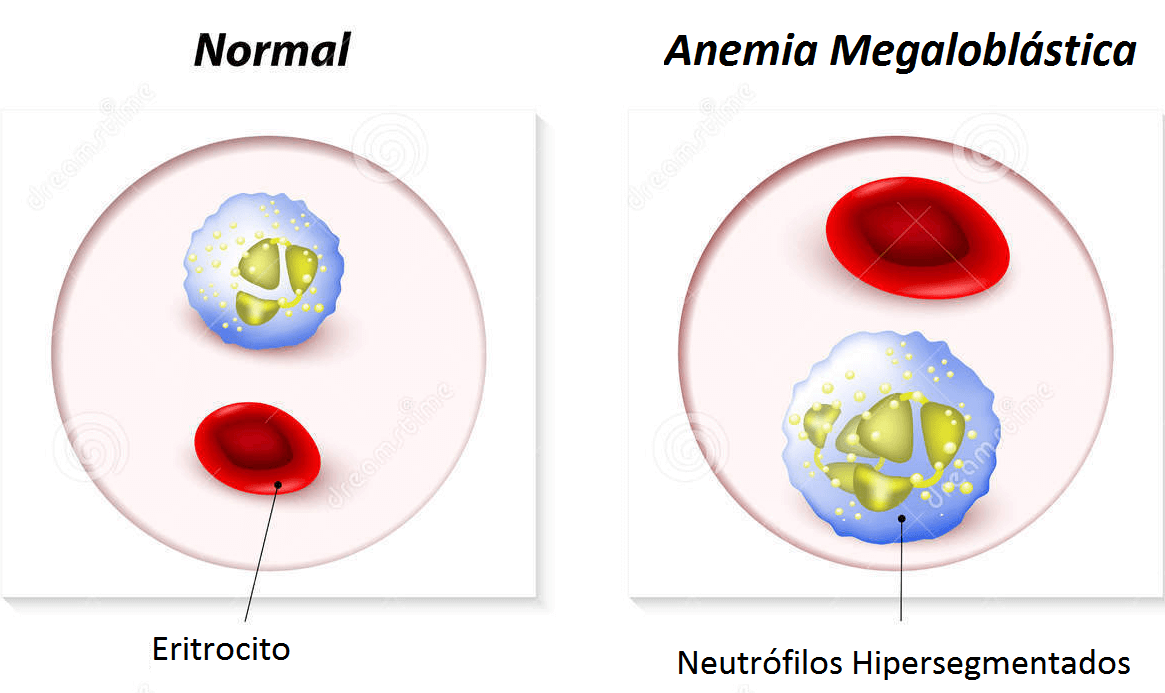
En muchos casos, las anemias hemolíticas son intensas, con concentraciones de hemoglobina <6 g/dl. La esferocitosis y la policromasia son importantes y más del 50% de los eritrocitos circulantes son reticulocitos, con presencia en ocasiones de hematíes nucleados principio del episodio. Es habitual encontrar leucocitosis. El recuento plaquetario suele ser normal, pero a veces se observa concomitantemente una púrpura trombocitopénica inmunitaria (síndrome de Evans). El pronóstico de los pacientes con síndrome de Evans es reservado, pues muchos padecen una enfermedad crónica que puede ser un LED o un trastorno linfoproliferativo autoinmunitario.
Tratamiento
Las transfusiones sólo aportan un beneficio transitorio pero pueden ser necesarias en un principio debido a la intensidad de la anemia y en tanto se espera el efecto de otros tratamientos. A veces resulta muy difícil encontrar sangre compatible; debe elegirse una sangre cuyos hematíes dan la reacción in vitro menos positiva con la técnica de Coombs. En ocasiones hay que administrar sangre que según las pruebas cruzadas es «incompatible». Sin transfusión, la morbilidad del lactante o del niño con anemia intensa puede ser muy grave y el paciente podría incluso morir.
Cuando la enfermedad es leve y la hemólisis está compensada, no se precisa tratamiento alguno. Si la hemólisis es intensa y provoca una anemia grave o sintomática, deberá iniciarse la administración de glucocorticoides. Éstos reducen la velocidad de la hemólisis porque bloquean la función de los macrófagos al disminuir la expresión del receptor Fc, reducen la producción de autoanticuerpos y, quizá, potencian la separación entre los anticuerpos y los eritrocitos. La posología a administrar es de 2 mg/kg/24 horas de prednisona o equivalente.
Evaluación y pronóstico
La gravedad de la forma aguda de las anemias hemolíticas autoinmunitaria idiopática de la infancia es variable pero el trastorno es autolimitado y la mortalidad secundaria a una anemia intratable es rara. Alrededor del 30% de los pacientes presentan hemólisis crónica, asociada a menudo a una enfermedad subyacente como LED, linfoma o leucemia. La presencia de anticuerpos antifosfolípido en los pacientes adultos con hemólisis inmunitaria predispone a la trombosis. En los casos crónicos, la mortalidad depende de la enfermedad primaria.
Anemias hemolíticas asociadas a anticuerpos fríos
Los anticuerpos «fríos» son anticuerpos antieritrocitarios que son más activos a temperaturas bajas y provocan su aglutinación por debajo de 37 °C se llaman anticuerpos «fríos». En su mayoría son de la clase IgM y para ser activos necesitan de la participación del complemento. El intervalo de temperatura al que producen aglutinación de los hematíes se denomina amplitud térmica. Una mayor amplitud térmica implicará hemólisis ante una exposición a un ambiente frío menos intensa. Los títulos de anticuerpos más elevados corresponden a los casos de mayor amplitud térmica.
Enfermedad aglutinas frías
Los anticuerpos fríos suelen ser específicos de oligosacáridos antigénicos del sistema I/i. Puede manifestarse como una enfermedad por aglutininas frías idiopática o primaria, de forma secundaria a infecciones por ejemplo por Mycoplasma pneumoniae o virus Epstein-Barr, o asociados a trastornos linfoproliferativos. Tras la infección por M. pneumoniae, los niveles de anti-I pueden aumentar de manera considerable y a veces se miden incrementos enormes, con títulos ≥1:30.000.
El anticuerpo es específico para el antígeno I y, por tanto, reacciona mal con los eritrocitos del cordón umbilical, que poseen sobre todo antígeno i y concentraciones bajas de I. Los pacientes con mononucleosis infecciosa presentan en ocasiones una enfermedad por aglutininas frías y sus anticuerpos suelen tener especificidad i. Este anticuerpo produce menos hemólisis en los adultos que en los niños, debido a que los eritrocitos de los adultos poseen menos moléculas i. Los hematíes se aglutinan espontáneamente con el frío y en las extensiones de sangre se encuentran agregados eritrocitarios. El volumen corpuscular medio puede parecer erróneamente alto debido a la aglutinación celular. La gravedad de la hemólisis depende de la amplitud térmica del anticuerpo, que a su vez depende en parte del título de anticuerpos IgM.
Cuando los títulos de anticuerpos fríos son muy altos y su actividad se desarrolla a temperaturas parecidas a la temperatura corporal normal, puede producirse una hemólisis intravascular con hemoglobinemia y hemoglobinuria, que aumenta cuando el paciente se expone al frío (temperatura exterior o alimentos ingeridos). Cada molécula de IgM puede activar una molécula de C1, por lo que en la enfermedad por aglutininas frías se encuentra gran cantidad de complemento sobre los hematíes. Estos hematíes sensibilizados pueden sufrir lisis intravascular por el complemento o pueden ser destruidos en el hígado y en el bazo.
La enfermedad por aglutininas frías es menos frecuente en los niños que en los adultos y en la mayoría de los casos se manifiesta por un episodio agudo y autolimitado de hemólisis. Los glucocorticoides son mucho menos eficaces en esta enfermedad que en la producida por anticuerpos calientes. Los pacientes deben evitar la exposición al frío y la enfermedad subyacente, si existe, debe ser tratada. En los casos poco frecuentes de hemólisis grave, el tratamiento consiste en inmunosupresión y plasmaféresis. Existen varias descripciones de tratamiento satisfactorio de la enfermedad por aglutininas frías con el anticuerpo monoclonal rituximab, que produce un eficaz agotamiento de los linfocitos B. La esplenectomía no es útil en esta enfermedad.
Hemolisis por fragmentación
En las anemias hemolíticas, la destrucción de los eritrocitos puede deberse a una lesión mecánica producida cuando la célula atraviesa un lecho vascular alterado. El daño puede ser microvascular, cuando la fibrina desgarra a los hematíes en los capilares durante una coagulación intravascular o cuando la enfermedad renovascular acompaña al síndrome hemolítico urémico o a la púrpura trombótica trombocitopénica.
En el síndrome de Kasabach-Merritt (hemangioma gigante y trombocitopenia; o cuando una prótesis valvular cardíaca se epiteliza mal, los vasos afectados son de mayor calibre. La extensión de sangre revela muchos «esquistocitos» o células fragmentadas, junto a una policromatofilia debida a la reticulocitosis (v. fig. 458-3F). La deficiencia secundaria de hierro puede complicar la hemólisis intravascular por pérdida urinaria del metal con la hemoglobina y la hemosiderina.
El tratamiento debe ir dirigido a corregir la alteración subyacente y el pronóstico depende de la efectividad de las medidas tomadas. El beneficio de las transfusiones es transitorio porque las células transfundidas se destruyen con igual rapidez que las producidas por el paciente.
-Lesión térmica. Las quemaduras extensas pueden lesionar de forma directa los eritrocitos y provocar una hemólisis que se traduce en la formación de esferocitos. La pérdida de sangre y la supresión medular pueden contribuir a la anemia y obligan a administrar transfusiones de sangre. Se ha utilizado la eritropoyetina (EPO) para corregir el trastorno de la producción de hematíes.
-Nefropatías. La anemia de la uremia es multifactorial. La producción de EPO puede disminuir, mientras que los metabolitos tóxicos inhiben la médula ósea. Además, la esperanza de vida de los hematíes suele ser corta debido a la retención de metabolitos y a la acidemia orgánica. El uso de EPO en las nefropatías crónicas ha reducido de manera notable la necesidad de transfusiones de sangre.
Hepatopatías. La alteración del cociente entre el colesterol y los fosfolípidos plasmáticos puede modificar la composición de la membrana eritrocitaria y acortar la esperanza de vida de los hematíes. En las extensiones de sangre de algunos pacientes con hepatopatías pueden encontrarse muchos eritrocitos en diana, mientras que en otros predominan las células espiculadas.
-Toxinas y Venenos. La sepsis bacteriana por Haemophilus influenzae, estafilococos y estreptococos puede complicarse con una hemólisis acompañante. En las infecciones por «clostridium» se describe una anemia especialmente grave, debida a la toxina hemolítica de estos microorganismos. En la extensión de sangre puede verse un gran número de esferocitos. Tras picaduras de distintas serpientes, por ejemplo cobras, víboras y serpientes de cascabel, que tienen fosfolipasas en su veneno, puede producirse una hemólisis esferocítica. Las picaduras de un gran número de insectos como avispas y abejas también provocan hemólisis esferocítica por un mecanismo similar .
Enfermedad de Wilson. La primera manifestación de una enfermedad de Wilson puede ser un episodio agudo y autolimitado de anemias hemolíticas que preceda en años a la aparición de los síntomas hepáticos o neurológicos. Parece que esta anemia se debe a los efectos tóxicos del cobre libre sobre la membrana eritrocitaria. A menudo (aunque no siempre) se encuentra en la extensión de sangre un gran número de esferocitos y la prueba de Coombs es negativa. Dado que un diagnóstico precoz de la enfermedad de Wilson permite su tratamiento profiláctico con penicilamina, que evita las manifestaciones hepáticas y neurológicas, es importante hacer una valoración correcta de este tipo raro de hemólisis.
Te interesara leer: 6 Tipos De Anemias Más Frecuentes, Síntomas Y Tratamiento!
.
¿Vulnera este post tus derechos? Pincha aquí.
Creado: