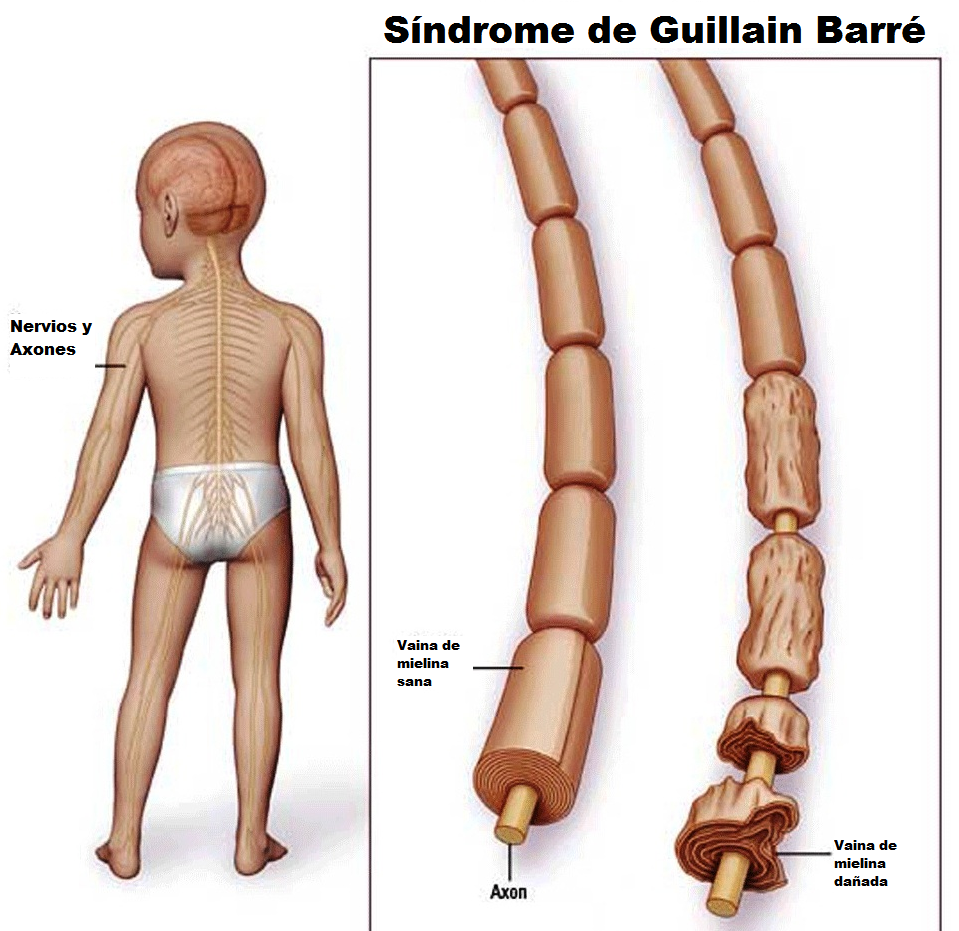
Signos y síntomas
La parálisis suele aparecer aproximadamente 10 días después de una infección vírica inespecífica. La infección original puede haber causado sólo síntomas gastrointestinales (especialmente por Campylobacter jejuni, pero también por Helicobacter pylori) o infección de las vías respiratorias (especialmente por Mycoplasma pneumoniae).
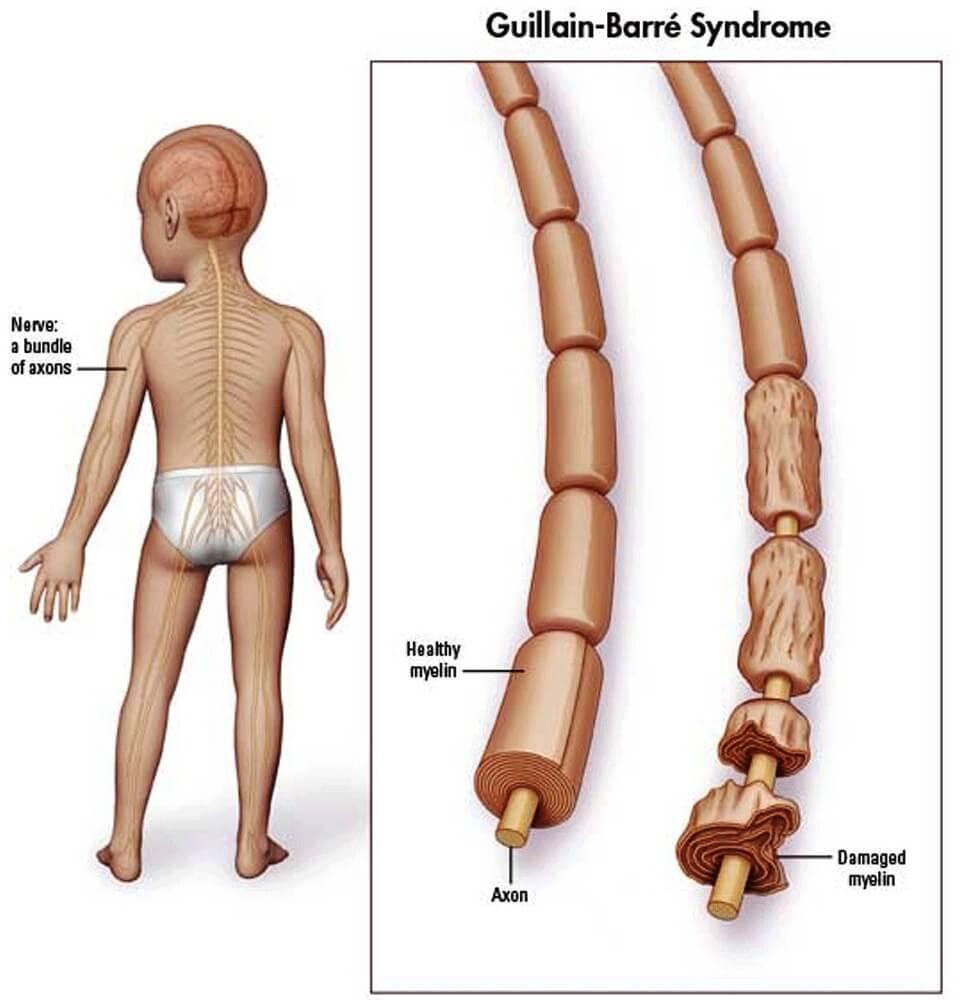
El virus del Oeste del Nilo también puede producir un cuadro parecido al síndrome de Guillain Barré, pero con mayor frecuencia origina un proceso similar a una enfermedad de neurona motora o poliomielitis. Se ha reportado síndrome de Guillain Barré tras la administración de vacunas frente a la rabia, gripe, polio (oral) y posiblemente con la vacuna conjugada del meningococo.
Debilidad Osteomuscular
La debilidad muscular suele comenzar en las extremidades inferiores y afecta progresivamente al tronco, los miembros superiores y finalmente a los músculos bulbares, un patrón conocido como parálisis ascendente de Landry. Los músculos proximales y distales están afectados de forma relativamente simétrica, pero en el 9% de los pacientes se encuentra cierta asimetría.
El inicio es gradual y progresivo durante días o semanas. En fases iniciales puede haber dolor muscular espontáneo o tras la palpación, sobre todo en los casos de comienzo brusco. Los niños afectados se encuentran irritables. La debilidad muscular puede progresar hasta la incapacidad o negativa a caminar y después puede causar hasta una tetraplejía flácida.
Afectación del bulbo raquídeo
En un 50% de los casos se produce afectación bulbar, que puede causar insuficiencia respiratoria. Con frecuencia, la disfagia y la debilidad muscular facial son signo de una insuficiencia respiratoria inminente.
Aprende que es la meningitis y sus principales causas
El síndrome de Miller-Fisher consiste en oftalmoplejía externa, ataxia y arreflexia. En algunos casos se observa edema de papila, pero no hay disminución de agudeza visual clínicamente. En el 20% de los casos se producen incontinencia o retención urinaria, pero suelen ser transitorias.
El síndrome de Miller-Fisher es superponible a la encefalitis troncoencefálica de Bickerstaff, que también muestra muchas características del síndrome de Guillain Barré con afectación de la neurona motora inferior y es posible que se trate de la misma enfermedad.
Los reflejos de estiramiento muscular desaparecen, normalmente de manera precoz durante la evolución, pero en ocasiones están conservados hasta estadios tardíos. Esta variabilidad puede causar confusión cuando se intenta realizar un diagnóstico precoz. El sistema nervioso autónomo también puede estar afectado en algunos casos.
Aparecen labilidad de la presión arterial y la frecuencia cardíaca, hipotensión postural, episodios de bradicardia profunda y en ocasiones asistolia. La monitorización cardiovascular es importante. Algunos pacientes requieren la implantación de un marcapasos cardíaco venoso transitorio.
La polirradiculoneuropatía crónica recidivante (en ocasiones denominada polirradiculoneuropatía desmielinizante inflamatoria crónica) lapolirradiculoneuropatía crónica no remitente son variantes crónicas del síndrome de Guillain Barré que recurren intermitentemente o que no mejoran durante un período de meses o años. Cerca del 7% de los niños con síndrome de Guillain Barré sufren una recaída aguda. Los pacientes suelen presentar debilidad muscular grave y pueden tener una tetraplejía flácida con o sin afectación de los músculos bulbares y respiratorios.
El síndrome de Guillain Barré congénito se describe en muy pocos casos y se caracteriza por hipotonía, debilidad muscular y arreflexia generalizadaen un recién nacido que cumple todos los criterios electrofisiológicos y del líquido cefalorraquídeo (LCR) en ausencia de enfermedad neuromuscular materna.
Puede no requerir tratamiento y existe una mejoría gradual durante los primeros pocos meses, sin evidencia de enfermedad residual durante el primer año de vida. En un caso, la madre tenía una colitis ulcerosa tratada con prednisona y mesalamina desde el 7.º mes de gestación hasta el parto a término.
Exámenes de laboratorio
El estudio del LCR es esencial para el diagnóstico. Las proteínas en el LCR están elevadas hasta más de dos veces por encima del límite superior normal, el nivel de glucosa es normal y no existe pleocitosis. Se encuentran menos de 10 leucocitos/mm3. Los resultados de los cultivos bacterianos son negativos y en los cultivos virales no se suelen aislar virus específicos.
Conoce los 7 trastornos de las neuronas motoras mas frecuentes
La disociación entre la elevación de las proteínas en el LCR y la ausencia de respuesta celular en un paciente con una polineuropatía aguda o subaguda es diagnóstica del síndrome de Guillain Barré. La velocidad de conducción nerviosa motora está muy retrasada, y en muchos casos el tiempo de latencia de los nervios sensitivos está aumentado. El electromiograma muestra signos de denervación aguda. La creatina cinasa (CK) sérica es normal o puede estar ligeramente elevada.
Las pruebas serológicas para demostrar la infección por Campylobacter y Helicobacter ayudan a establecer la causa cuando son positivas, pero no alteran el curso del tratamiento. Los resultados de los cultivos de heces casi siempre son negativos porque la infección es autolimitada, sólo dura aproximadamente 3 días y la neuropatía es posterior a la gastroenteritis aguda.
Tratamiento
Los pacientes en estadios precoces de esta enfermedad aguda deben ingresarse en el hospital para su observación porque la parálisis ascendente puede afectar rápidamente a los músculos respiratorios. Durante las siguientes 24 horas.
Los pacientes con progresión lenta pueden someterse simplemente a observación para su estabilización hasta la remisión espontánea sin tratamiento. La parálisis ascendente rápidamente progresiva se trata con inmunoglobulina intravenosa (IGIV) administrada durante 2, 3 o 5 días. La administración combinada de inmunoglobulina e interferón es eficaz en algunos pacientes.
El tratamiento de soporte, como la ventilación asistida, la prevención de las úlceras por decúbito en los niños con tetraplejía flácida y el tratamiento de las infecciones bacterianas secundarias, es importante. La polirradiculoneuropatía crónica recidivante o neuropatía crónica no remitente también se trata con IGIV.
La plasmaféresis, necesaria en ocasiones hasta 10 veces al día, es una alternativa. La remisión en estos casos puede ser mantenida, pero las recaídas pueden aparecer al cabo de días, semanas o incluso después de muchos meses y generalmente responden a otro ciclo de plasmaféresis. Los esteroides y los fármacos inmunosupresores constituyen otra alternativa, pero su eficacia es menos predecible. La metilprednisolona, a altas dosis, administrada por vía intravenosa es eficaz en algunos casos.
El pronóstico en las formas crónicas del síndrome de Guillain-Barré es más reservado que en la forma aguda y muchos pacientes permanecen con discapacidades residuales significativas. Incluso cuando se detecta una infección por Campylobacter jejuni mediante los cultivos de heces o las pruebas serológicas, el tratamiento de la infección no es necesario porque es una infección autolimitada y la utilización de antibióticos no altera la evolución de la polineuropatía.
Pronostico
El curso clínico suele ser benigno y la recuperación espontánea comienza a las 2-3 semanas. La mayoría de los pacientes recuperan completamente la fuerza muscular, aunque algunos presentan debilidad residual. Los reflejos miotáticos generalmente son la última función que se recupera. La mejoría clínica habitualmente sigue un gradiente de dirección inverso al de la afectación, resolviéndose antes la debilidad de los músculos bulbares que la de los músculos de las extremidades.
La afectación de la musculatura bulbar y respiratoria puede provocar la muerte si el síndrome no se reconoce y se trata. Aunque el pronóstico suele ser bueno y la mayoria de los niños se recuperan por completo, hay tres signos clínicos que pueden predecir una evolución con secuelas: afectación de nervios craneales, intubación y máxima afectación en el momento de la presentación. La presencia de bloqueos de conducción en el estudio neurofisiológico es indicativa de buen pronóstico.
El seguiento a largo plazo de pacientes que se han recuperado de un síndrome de Guillain Barré revela que alguno de ellos puede tener pérdidas axonales permanentes, con o sin signos residuales de neuropatía crónica. La fatigabilidad precoz es uno de los síntomas crónicos más frecuentes, pero la fatigabilidad muscular no es tan rápida como en la miastenia grave. Los pacientes con la forma axonal del síndrome de Guillain Barré mejoran lentamente durante los primeros 6 meses, pudiendo eventualmente caminar, aunque algunos requieren años para recuperarse. Los estudios neurofisiológicos, EMG y VCN no predicen necesariamente la evolución a largo plazo.
La entrada 4 Síntomas del Síndrome de Guillain Barré aparece primero en Mega Medico.
¿Vulnera este post tus derechos? Pincha aquí.
Creado: