La policitemia o poliglobulia se define como un aumento en la masa eritrocitaria. Se caracteriza por un incremento del número de hematíes y/o de la cantidad de hemoglobina por unidad de volumen de sangre. El parámetro hematológico mas apropiado para su valoración es el hematocrito, debiéndose sospechar poliglobulia cuando este se sitúa de forma mantenida dos desviaciones estándar por encima de la media normal: 52% en varones y al 48% en mujeres. En caso de utilizarse la hemoglobina se debe sospechar poliglobulia cuando la hemoglobina es mayor de 18,5 g/dl en varones y de 16,5 g/dl en mujeres.
CLASIFICACIÓN Y ETIOLOGÍA
Es importante diferenciar si la poliglobulia es absoluta, con un aumento real de la masa eritrocitaria, o relativa, en la que hay un incremento de la concentración de hematíes por una pérdida del volumen plasmático, pero la masa eritrocitaria es normal.
Poliglobulia relativa o ficticia
Es la poliglobulia que se produce por disminución del volumen plasmático, con un aumento relativo en la concentración de hematíes. Aparece en procesos que causan deshidratación, como vómitos de repetición, diarrea grave, uso excesivo de laxantes y diuréticos, etc. También se observa en el síndrome de Gaisböck -poliglobulia aparente o de estrés-, cuadro de etiología desconocida que afecta a varones de mediana edad con HTA, obesidad y estados de ansiedad.
Poliglobulia absoluta
Aquella poliglobulia con un aumento real de la masa eritrocitaria total. A su vez se clasifica en:
Policitemia primaria
La policitemia vera es una neoplasia mieoloproliferativa causadas por una mutación adquirida (JAK 2 V617F -presente en el 90% de los casos- u otra mutación comparable, JAK 2 exon 12), que conduce a una alteración intrínseca de la eritropoyesis medular, y que se caracteriza por un incremento en la producción de los progenitores de los glóbulos rojos, independiente de los mecanismos que regulan normalmente la eritropoyesis, siendo las concentraciones de eritropoyetina (EPO) normales o bajas. También dentro de este grupo se incluyen algunas variantes familiares de carácter hereditario poco frecuentes.
Eritrocitosis idiopática
Se ha utilizado este término para clasificar a los pacientes con policitemia primaria que no cumplen los criterios convencionales para el diagnóstico de policitemia vera, incluyendo la negatividad para la mutación JAK 2 V617F y JAK 2 exon 12.
Policitemia secundaria
Policitemia causada por un aumento del factor estimulante de la eritropoyesis (EPO), siendo normales los precursores de los hematíes de la médula ósea. La producción de EPO puede ser:
Debido a una respuesta fisiológica a la hipoxia crónica por EPOC, hipoventilación alveolar, cardiopatías congénitas con shunt derecha-izquierda, síndrome de Pickwick (obesos), aumento de carboxihemoglobina (fumadores), permanencia en grandes alturas, descenso congénito del 2,3 DPG, intoxicación por cobalto, hemoglobinas con aumento de la afinidad por el oxígeno, síndrome de apnea del sueño, defectos neurológicos (disfunciones del centro respiratorio), etc.
Resultado de una secreción anormal de EPO, en el caso de lesiones renales, como por ejemplo, estenosis arteria renal, hidronefrosis, poliquistosis renal, glomerulonefritis, trasplante renal, o en tumores secretores como el carcinoma renal, carcinoma hepatocelular y hemangioblastoma cerebeloso, tumores androgénicos, mioma uterino, cáncer de ovario, adenoma suprarrenal, y en otros trastornos como hepatitis, cirrosis, tratamientoon andrógenos, etc.
Poliglobulia esencial o idiopática.
Poliglobulia en la que no se identifica una causa primaria ni secundaria. Un 5-10% terminan desarrollando una policitemia vera con el paso de los años.
Policitemia combinada
Policitemia caracterizada por tener un aumento de la masa eritrocitaria, así como un volumen reducido de plasma, una combinación más comúnmente vista en los fumadores ?policitemia de los fumadores-.
Policitemia inaparente.
Policitemia caracterizada por una masa eritrociataria aumentada con cifras de hematocrito y hemoglobina normales. Solo se puede detectar a través de estudios de la masa eritrocitaria.
CLÍNICA
La poliglobulia se suele presentar como un cuadro insidioso y progresivo con síntomas en general inespecíficos, como astenia, sensación de mareo o vértigo, acufenos, cefalea y epistaxis. Las poliglobulias secundarias además presentará síntomas relacionados con el proceso subyacente, como es una enfermedad cardíaca, pulmonar o renal.
En los pacientes con poliglobulia la incidencia de complicaciones trombóticas es mayor. Los episodios más frecuentes son accidentes cerebrovasculares (ACVA), infarto de miocardio, trombosis venosa profunda y tromboembolismo pulmonar. Además, los pacientes con policitemia vera pueden presentar hemorragias en el tracto gastrointestinal, por un descenso relativo de los factores de la coagulación y en algunos casos trastornos funcionale de las plaquetas, y una mayor incidencia de úlceras pépticas y varices esofágicas secundarias a hipertensión portal.
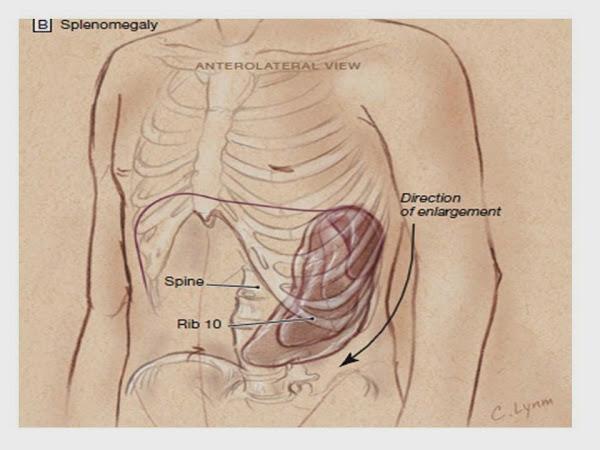
En la exploración física los signos más característicos son la cianosis rubicunda, plétora de predominio facial e inyección conjuntival. Los pacientes con policitemia vera presentan esplenomegalia en el 60% de los casos y hepatomegalia en el 40%. En los casos de poliglobulia de estrés (síndrome de Gaisböck) destacan la obesidad y la hipertensión arterial (HTA).
Debido a la activa renovación de las células hemáticas se produce una hiperuricemia y son frecuentes en estos pacientes los episodios de gota.
DIAGNÓSTICO
Anamnesis
En muchas ocasiones la poliglobulia se detecta de forma casual a partir de una analítica, ya que el paciente está asintomático. En caso de presentar alguno/s de los síntomas y/o signos ya referidos que hagan sospechar una posible poliglobulia, se deben realizar una anamnesis y exploración dirigidas, así como las pruebas complementarias necesarias para su confirmación.
Es importante conocer el consumo de tabaco y de fármacos (diuréticos, laxantes) que puedan provocar deshidratación, la existencia de HTA, enfermedades cardíacas, pulmonares, renales y hepáticas y los antecedentes familiares de poliglobulia.
Exploraciones complementarias
Hemograma. Es fundamental para confirmar que existe un aumento de la concentración de hematíes, con valores de hematocrito y/o hemoglobina elevados. Son importantes las cifras de leucocitos y plaquetas, ya que suelen estar elevadas en la policitemia vera al ser un síndrome mieloproliferativo y también resulta de interés la extensión de sangre periférica para estudiar la morfología de los hematíes.
Bioquímica. Incluye el ácido úrico, lactatodeshidrogenasa, fosfatasa alcalina y vitamina B12. Todos ellos están elevados en la policitemia vera y son normales en las otras poliglobulias. Otras exploraciones incluyen estudio del metabolismo del hierro, aclaramineto de creatinina, pruebas hepáticas, y acido úrico
Radiografía de tórax y electrocardiograma. Para descartar la presencia de enfermedad cardíaca y pulmonar.
En una segunda fase se deben de realizar otras exploraciones en función de los datos obtenidos con las pruebas anteriores
Ecografía abdominal. Permite confirmar la existencia de esplenomegalia que orienta hacia una policitemia vera y descartar la presencia de enfermedad renal y hepática.
Gasometría arterial basal. A valorar en pacientes fumadores. Ayuda a descartar la hipoxia como causa de poliglobulia.
Eritropoyetina sérica (EPO). Ayuda a diferenciar entre una poliglobulia secundaria y una policitemia vera en los casos dudosos.
Volumen sanguíneo. Se cuantifica la masa eritrocitaria total, por técnicas de dilución con isótopos radiactivos como el cromo 515, y el volumen plasmático para diferenciar entre una poliglobulia absoluta y una relativa.
Otras pruebas adicionales y específicas en función de la orientación diagnóstica a partir de los estudios previos, incluyen: estudio de PCR alelo específico para la mutación V617F del gen JAK2, curva de disociación del O2 (P50), test de función respiratoria, ecocardiograma, estudio genético del receptor de la eritropoyetina, etc.
Biopsia de médula ósea. Está indicado ante la sospecha de policitemia vera para confirmar el diagnóstico e incluye punción-aspiración y biopsia.
POLICITEMIA VERA
La policitemia vera es una neoplasia mieloproliferativa caracterizada por un incremento en la producción de eritrocitos, independiente de los mecanismos que regulan normalmente la eritropoyesis. Su incidencia anual es de aproximadamente 0.02-2.8 casos por cada 100.000 habitantes. Los síntomas son consecuencia de la proliferación celular excesiva -cefalea, astenia, alteraciones visuales, disnea, prurito, sudoración, inyección conjuntival, hepato-esplenomegalia, e hipertensión arterial. Las complicaciones trombóticas arteriales y venosas -ocurren en el 40% de los pacientes- son la principal causa de morbimortalidad. También las complicaciones hemorrágicas son frecuentes -30-40% de los pacientes-, siendo características la epistaxis, las hemorragias retinianas, gastrointestinales y cerebrales.
El diagnóstico se basa en el cumplimiento de los siguientes criterios establecidos por la OMS, requiriéndose la existencia de los dos criterios mayores y uno menor, o el primer criterio mayor y dos criterios menores:
Criterios Mayores
1. Hemoglobina > 18.5 g/dL (16.5 g/dL en mujeres)
2. Existencia de JAK 2 V617F -presente en el 90% de los casos- u otra mutación comparable (JAK 2 exon 12)
Criterios Menores
1. Biopsia de médula ósea hipercelular.
2. Eritropoyetina sérica por debajo del límite normal
3. Formación de colonias eritroides endógenas in vitro
Tabla 1.- Criterios diagnósticos de policitemia vera
TRATAMIENTO
El tratamiento específico de las poliglobulias es variable en función de su etiología. Los casos relacionados con deshidratación o tabaquismo remiten al desaparecer la causa subyacente. Los procesos secundarios a enfermedad cardiopulmonar, renal o hepática requieren tratamiento de la enfermedad de base. La policitemia vera, al ser una neoplasia, precisa generalmente tratamiento mielosupresor asociado a flebotomías. En su tratamiento se emplean:
Ácido acetilsalicílico, 100 mg/24 h: indicado en todos los casos.
Flebotomía: indicada en todos los pacientes con hematocrito superior al 54% -o inferior en presencia de otros factores de riesgo trombótico-, hasta obtener valores por debajo del 45% y para mantenerlo entre 42-45%.
Agentes mielosupresores: Indicados por intolerancia a las flebotomías, por la presencia de mieloproliferación -manifestada por esplenomegalia refractaria o un recuento leucocitario o plaquetario elevado-, o por la existencia de un alto riesgo trombótico. La hidroxiurea es actualmente el agente mielosupresor de elección, proporcionando un control adecuado de las enfermedades en la mayoría de los pacientes. Sin embargo, persiste la controversia sobre si aumenta el riesgo de transformación leucémica a largo plazo -10-15 años-. El interferón alfa no es un fármaco de primera elección pero puede ser útil en pacientes que han recaído con hidroxiurea, mujeres embarazadas, y en personas menores de 50 años. El busulfán y el fosforo 32 pueden estar especialmente indicados en pacientes mayores de 70 años. La anagrelida puede estar indicada para el tratamiento de la trombocitosis en pacientes refractarios a la hidroxiurea.
BIBLIOGRAFÍA RECOMENDADA
Aitchison R, Russell N. Smoking--a major cause of polycythaemia. J R Soc Med 1988; 81:89.
Bacon BR, Rothman SA, Ricanati ES, Rashad FA. Renal artery stenosis with erythrocytosis after renal transplantation. Arch Intern Med 1980; 140:1206.
Bernard PJ. Measurement of red-cell and plasma volumes. Nouv Rev Fr Hematol 1994; 36:155.
Birgegård G, Wide L. Serum erythropoietin in the diagnosis of polycythaemia and after phlebotomy treatment. Br J Haematol 1992; 81:603.
Brown SM, Gilbert HS, Krauss S, Wasserman LR. Spurious (relative) polycythemia: a nonexistent disease. Am J Med 1971; 50:200.
Chagnac A, Zevin D, Weinstein T, et al. Erythrocytosis associated with renal artery thrombosis in a patient with polycystic kidney disease on hemodialysis. Acta Haematol 1990; 84:40.
Da Silva JL, Lacombe C, Bruneval P, et al. Tumor cells are the site of erythropoietin synthesis in human renal cancers associated with polycythemia. Blood 1990; 75:577.
Distelhorst CW, Wagner DS, Goldwasser E, Adamson JW. Autosomal Dominant familial erythrocytosis due to autonomous erythropoietin production. Blood 1981; 58:1155.
Drénou B, Le Tulzo Y, Caulet-Maugendre S, et al. Pheochromocytoma and secondary erythrocytosis: role of tumour erythropoietin secretion. Nouv Rev Fr Hematol 1995; 37:197.
Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169.
Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151.
Gaston RS, Julian BA, Curtis JJ. Posttransplant erythrocytosis: an enigma revisited. Am J Kidney Dis 1994; 24:1.
Gregg XT, Prchal JT. Erythropoietin receptor mutations and human disease. Semin Hematol 1997; 34:70.
Holme S, Elfath MD, Heaton A, et al. Prediction of red cell and blood volumes distribution by various nomograms: do current nomograms overestimate? Transfusion 2008; 48:910.
Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A 1996; 93:10595.
Johansson PL, Safai-Kutti S, Kutti J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: a study of patients with polycythaemia vera and apparent polycythaemia. Br J Haematol 2005; 129:701.
Juvonen E, Ikkala E, Fyhrquist F, Ruutu T. Autosomal dominant erythrocytosis caused by increased sensitivity to erythropoietin. Blood 1991; 78:3066.
Kew MC, Fisher JW. Serum erythropoietin concentrations in patients with hepatocellular carcinoma. Cancer 1986; 58:2485.
Kralovics R, Indrak K, Stopka T, et al. Two new EPO receptor mutations: truncated EPO receptors are most frequently associated with primary familial and congenital polycythemias. Blood 1997; 90:2057.
Lamy T, Devillers A, Bernard M, et al. Inapparent polycythemia vera: an unrecognized diagnosis. Am J Med 1997; 102:14.
Lanne JS, Dumortier J, Hervieu V, et al. Polycythemia and elevated serum erythropoietin associated with a liver haemangioma. Gastroenterol Clin Biol 2010; 34:629.
LAWRENCE JH, BERLIN NI. Relative polycythemia; the polycythemia of stress. Yale J Biol Med 1952; 24:498.
LevGur M, Levie MD. The myomatous erythrocytosis syndrome: a review. Obstet Gynecol 1995; 86:1026.
McMullin MF, Bareford D, Campbell P, et al. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br J Haematol 2005; 130:174.
Messinezy M, Sawyer B, Westwood NB, Pearson TC. Idiopathic erythrocytosis--additional new study techniques suggest a heterogenous group. Eur J Haematol 1994; 53:163.
Messinezy M, Westwood NB, El-Hemaidi I, et al. Serum erythropoietin values in erythrocytoses and in primary thrombocythaemia. Br J Haematol 2002; 117:47.
Najean Y, Schlageter MH, Toubert ME, Podgorniak MP. Radioimmunoassay of immunoreactive erythropoietin as a clinical tool for the classification of polycythaemias. Nouv Rev Fr Hematol 1990; 32:237.
Nielsen S, Rødbro P. Validity of rapid estimation of erythrocyte volume in the diagnosis of polycytemia vera. Eur J Nucl Med 1989; 15:32.
Pearson TC, Guthrie DL, Simpson J, et al. Interpretation of measured red cell mass and plasma volume in adults: Expert Panel on Radionuclides of the International Council for Standardization in Haematology. Br J Haematol 1995; 89:748.
Pearson TC, Messinezy M. Investigation of patients with polycythaemia. Postgrad Med J 1996; 72:519.
Prchal JT, Sokol L. "Benign erythrocytosis" and other familial and congenital polycythemias. Eur J Haematol 1996; 57:263.
Remacha AF, Montserrat I, Santamaria A, et al. Serum erythropoietin in the diagnosis of polycythemia vera. A follow-up study. Haematologica 1997; 82:406.
Rossi D, Cortini F, Deambrogi C, et al. Usefulness of JAK2V617F mutation in distinguishing idiopathic erythrocytosis from polycythemia vera. Leuk Res 2007; 31:97.
Ruggeri M, Tosetto A, Frezzato M, Rodeghiero F. The rate of progression to polycythemia vera or essential thrombocythemia in patients with erythrocytosis or thrombocytosis. Ann Intern Med 2003; 139:470.
Schmidt W, Prommer N. The optimised CO-rebreathing method: a new tool to determine total haemoglobin mass routinely. Eur J Appl Physiol 2005; 95:486.
Schramek A, Better OS, Adler O, et al. Hypertensive crisis, erythrocytosis, and uraemia due to renal-artery stenosis of kidney transplants. Lancet 1975; 1:70.
Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356:459.
Smith JR, Landaw SA. Smokers" polycythemia. N Engl J Med 1978; 298:6.
Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood 2002; 100:4272.
Spivak JL. The optimal management of polycythaemia vera. Br J Haematol 2002; 116:243.
Suzuki M, Takamizawa S, Nomaguchi K, et al. Erythropoietin synthesis by tumour tissues in a patient with uterine myoma and erythrocytosis. Br J Haematol 2001; 113:49.
Tefferi A. JAK2 mutations in polycythemia vera--molecular mechanisms and clinical applications. N Engl J Med 2007; 356:444.
Weinberg RS. In vitro erythropoiesis in polycythemia vera and other myeloproliferative disorders. Semin Hematol 1997; 34:64.
Wiesener MS, Seyfarth M, Warnecke C, et al. Paraneoplastic erythrocytosis associated with an inactivating point mutation of the von Hippel-Lindau gene in a renal cell carcinoma. Blood 2002; 99:3562.
¿Vulnera este post tus derechos? Pincha aquí.
Creado: