Los síndromes mieloproliferativos crónicos (SMP) son un grupo de trastornos hematológicos que se producen como consecuencia de una alteración clonal de la célula madre hematopoyética, dando lugar a la proliferación de una o más líneas celulares de la médula ósea. A diferencia de los síndromes mielodisplásicos, en los SMP esta proliferación se asocia a una maduración celular relativamente normal, que se manifiesta por un número elevado de granulocitos, hematíes y/o plaquetas en sangre periférica (SP), junto con hepato y/o esplenomegalia por metaplasia mieloide en estas vísceras, aparición en mayor o menor grado de fibrosis de la médula ósea, y en ocasiones evolución a leucemia aguda.
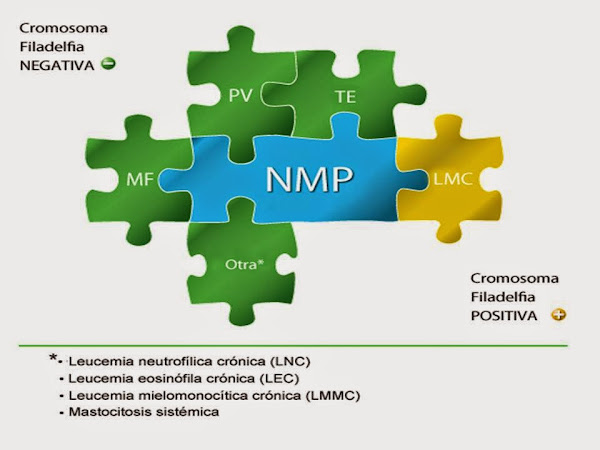
En 2008 la Organización Mundial de la Salud (OMS) propuso una nueva clasificación de los SMP, incorporando criterios clínicos, morfológicos y genéticos, que tiene implicaciones diagnósticas, pronosticas y terapéuticas, pasando a denominarse Neoplasias Mieloproliferativas (NMP), entre las que se incluyen las siguientes entidades: Leucemia Mieloide Crónica (LMC), Policitemia Vera (PV), Trombocitemia Esencial (TE), Mielofibrosis Primaria (MFP), Leucemia Neutrofílica Crónica (LNC), Leucemia Eosinofílica Crónica (LEC), Mastocitosis y Neoplasias Mieloproliferativas no clasificables (NMPNC).

Etiología
La etiología de las NMP es desconocida, aunque algunos casos se han relacionado con la exposición a radiaciones ionizantes y determinados disolventes orgánicos. Los primeros datos de un origen clonal de las NMP se descubrieron con los estudios de las isoenzimas de la glucosa-6-fosfatodeshidrogenasa. Posteriormente se descubrió que la LMC presenta una alteración citogenética específica, el cromosoma Filadelfia (Ph+), presente en el 95% de los casos. Más recientemente, el descubrimiento de la mutación JAK2 V617F, en un gen que se encuentra en la brazo corto del cromosoma 9, puso en evidencia una patogénesis común para las NMP con cromosoma Ph-. Esta mutación, que codifica a enzimas tirosina quinasas, responsables de la activación y crecimiento celular e independientes de los factores habituales del crecimiento celular, se encuentra en más del 90% de los pacientes con PV, en un 60% de los casos de TE y en aproximadamente el 50% de la MFP. También se han descrito otras alteraciones genéticas mas infrecuentes; como las del gen del receptor de la TPO (c-MPL) y la del gen del receptor del factor de crecimiento derivados de las plaquetas (PDGFR).

Leucemia mieloide crónica
La LMC es la NMP más frecuente, estimándose una incidencia anual de 1-2 casos por 100.000 habitantes. Típicamente se presenta de forma insidiosa, siendo los síntomas típicos de la enfermedad astenia, anorexia y pérdida de peso. Sin embargo, cerca de 40% de los pacientes están asintomáticos al inicio y su diagnóstico se basa únicamente en una analítica de rutina. En la exploración física destaca la existencia de hepatomegalia y esplenomegalia -presente en el 50-80% de los pacientes- que a veces puede ser gigante.En el hemograma destaca la presencia de leucocitosis, en torno a 50-100 × 109/l, a expensas de granulocitos y mielocitos, junto con basofilia y eosinofilia. La cifra de hemoglobina puede ser normal o estar ligeramente disminuida y el recuento de plaquetas suele estar elevado. En la bioquímica, la fosfatasa alcalina granulocitaria está disminuida en el 95% de los casos, los niveles séricos de vitamina B12, ácido úrico y LDH suelen estar elevados debido al alto turnover celular. La medula osea (MO) es hipercelular, con una marcada hiperplasia granulocítica a expensas de mielocitos y elementos maduros, con basofilia y eosinofilia. También es frecuente la hiperplasia megacariocítica. El porcentaje de blastos es < 5-10% y en un 30% de los casos se detecta fibrosis en grado variable. El estudio citogenético demuestra la existencia del cromosoma Ph + en el 95% de los casos, -se denomina cromosoma Ph al cromosoma 22 alterado, resultado de que una sección del cromosoma 9 y otra sección del cromosoma 22 se rompen e intercambian lugares, uniéndose en el cromosoma 22 y formando el oncogén BCR-ABL-, mientras que el resto de los pacientes presentan translocaciones mas extrañas que no pueden ser detectadas por análisis citogenéticos rutinarios, o presentan translocaciones complejas que involucran la participación de un tercer cromosoma. La presencia del cromosoma Ph no sólo en los precursores granulocíticos sino también en los eritrocitos, megacariocitos y linfocitos B -y posiblemente en los T- indica que el trastorno que origina la leucemia mieloide crónica radica en la célula madre (stem cell) común a todas las células hematopoyéticas. También hay que saber que existen casos de sujetos sanos que presentan cromosoma Ph+ sin desarrollar la enfermedad, por lo que se ha sugerido que en ellos la anormalidad cromosómica se origina en células progenitoras cuya descendencia está destinada a morir después de cierto número de divisiones celulares.
La historia natural de la LMC consta de tres fases: crónica, acelerada y blástica, o de trasformación en leucemia aguda terminal. La fase crónica se controla bien con el tratamiento y el paciente permanece asintomático durante años. La fase acelerada se caracteriza por el empeoramiento clínico, menor respuesta al tratamiento, esplenomegalia y leucocitosis progresivas y refractarias, basófilos en SP >20%, plaquetas > 1.000 × 109/l o < 100 × 109/l de forma persistente, y reaparición de la mieloproliferación, con 10-19% blastos en SP y/o en MO. Pocos meses después de la fase acelerada aparece la fase blástica o leucemia aguda terminal, con un incremento progresivo del número de blástos en SP y MO hasta superar el 20%. Dos tercios son de tipo mieloblástico y un tercio de tipo linfoblástico, siendo frecuente la aparición de clonas con anomalías citogenéticas, adicionales al cromosoma Ph, que se asocian a un peor pronóstico, y entra las que se incluyen doble Ph, i(17q), monosomía del 7, trisomía del 8 o pérdida del cromosoma Y.
El tratamiento de la LMC ha evolucionado notablemente. Históricamente la enfermedad presentaba una supervivencia de 2-3 años sin tratamiento, pasando a conseguirse, en tiempos relativamente recientes, una supervivencia de 4 años con busulfán e hidroxiurea, y de 6 años con tratamiento con interferón alfa y trasplante alogénico de células hematopoyéticas. En la actualidad, el tratamiento con los inhibidores de tirosincinasa (ITK), en especial el imatinib -ITK de primera generación-, han causado una verdadera revolución en el pronóstico de la LMC, con una tasa global de supervivencia a los 8 años del 85%, existiendo evidencias que sugiere que algunos pacientes pueden curarse. La aparición de nuevos agentes, como: dasatinib, nilotinib o bosutinib hacen más interesante el campo de tratamiento de la LMC, porque son más potentes y eficaces. El trasplante no ha desaparecido como opción terapéutica, pero se utiliza en forma más selecta y con métodos no mieloablativos.
Policitemia vera
La PV es una NMP caracterizada por un incremento en la producción de eritrocitos, independiente de los mecanismos que regulan normalmente la eritropoyesis. Su incidencia anual es de aproximadamente 0.02-2.8 casos por cada 100.000 habitantes. Los síntomas son consecuencia de la proliferación celular excesiva -cefalea, astenia, alteraciones visuales, disnea, prurito, sudoración, inyección conjuntival, hepato-esplenomegalia, e hipertensión arterial. Las complicaciones trombóticas arteriales y venosas -ocurren en el 40% de los pacientes- son la principal causa de morbimortalidad. También las complicaciones hemorrágicas son frecuentes -30-40% de los pacientes-, siendo características la epistaxis, las hemorragias retinianas, gastrointestinales y cerebrales.El diagnóstico se basa en el cumplimiento de los siguientes criterios establecidos por la OMS, requiriéndose la existencia de los dos criterios mayores y uno menor, o el primer criterio mayor y dos criterios menores:
? Criterios Mayores
1. Hemoglobina > 18.5 g/dL (16.5 g/dL en mujeres)
2. Existencia de JAK 2 V617F -presente en el 90% de los casos- u otra mutación comparable (JAK 2 exon 12)
? Criterios Menores
1. Biopsia de médula ósea hipercelular.
2. Eritropoyetina sérica por debajo del límite normal
3. Formación de colonias eritroides endógenas in vitro
La historia natural de la PV consta de tres fases: Fase inicial, fase policitémica y fase de agotamiento. La fase inicial es asintomática -sólo se detecta una eritrocitosis aislada y esplenomegalia, asociada o no a trombocitosis. La fase policitémica se asocia con aumento extremo de la masa eritrocítica y aparición de la clínica descrita. Finalmente la fase de agotamiento cursa con citopenias, mielofibrosis, hematopoyesis extramedular; es característica una reacción leucoeritroblástica con poiquilocitosis y dacriocitos en sangre periférica y esplenomegalia progresiva, debida a la hematopoyesis extramedular (metaplasia mieloide).
El pronóstico de los pacientes depende de la naturaleza y gravedad de las complicaciones que presenten en su evolución, así como de la duración de la fase crónica y de la evolución a mielofibrosis o leucemia aguda -la PV tiene una baja incidencia de trasformación en leucemia aguda, cuestionándose en la actualidad que la transformación blástica sea parte de la evolución natural de la PV, siendo posible que esté provocada, al menos en parte, por el tratamiento quimioterápico, especialmente el fosforo 32 y los agentes alquilantes-.
En su tratamiento se emplean:
Ácido acetilsalicílico, 100 mg/24 h: indicado en todos los casos.
Flebotomía: indicada en todos los pacientes con hematocrito superior al 54% -o inferior en presencia de otros factores de riesgo trombótico-, hasta obtener valores por debajo del 45% y para mantenerlo entre 42-45%.
Agentes mielosupresores: Indicados por intolerancia a las flebotomías, por la presencia de mieloproliferación -manifestada por esplenomegalia refractaria o un recuento leucocitario o plaquetario elevado-, o por la existencia de un alto riesgo trombótico. La hidroxiurea es actualmente el agente mielosupresor de elección, proporcionando un control adecuado de las enfermedades en la mayoría de los pacientes. Sin embargo, persiste la controversia sobre si aumenta el riesgo de transformación leucémica a largo plazo -10-15 años-. El interferón alfa no es un fármaco de primera elección pero puede ser útil en pacientes que han recaído con hidroxiurea, mujeres embarazadas, y en personas menores de 50 años. El busulfán y el fosforo 32 pueden estar especialmente indicados en pacientes mayores de 70 años. La anagrelida puede estar indicada para el tratamiento de la trombocitosis en pacientes refractarios a la hidroxiurea. Trombocitemia esencial
Se trata de una NMP caracterizada por un incremento persistente de la cifra de plaquetas con hiperplasia megacariocítica de la MO. Su incidencia anual aproximada es de 0,1-1,5 casos por cada 100.000 habitantes. Los síntomas son consecuencia son los trastornos tromboticos arteriales, mucho más frecuente que los venosos. Las manifestaciones tromboticas microvasculares dan lugar a eritromelalgia, -sensación de quemazón, dolor intenso y enrojecimiento de los dedos de las extremidades o de la planta del pie-. Las hemorragias son infrecuentes. También pueden darse complicaciones obstétricas como el aborto espontáneo, el retraso en el crecimiento fetal, la muerte intrauterina y la prematuridad.El diagnóstico de la TE se basa en los siguientes criterios:
Incremento sostenido de plaquetas igual o mayor de 450 X 109/l.
Incremento en el número de megacariocitos maduros en la biopsia de MO, sin alteraciones de serie roja o granulocítica.
Ausencia de criterios diagnósticos para PV, MFP o LCM, síndrome mielodisplásico (SMD) u otras NMP.
Presencia de JAK2V617F ?positivo en el 60% de los casos- ó MPL W515K/L.
Estudios recientes indican una supervivencia media que se acerca a 20 años siendo la transformación fibrótica o leucémica relativamente poco frecuente. El tratamiento debe decidirse tras evaluar el riesgo trombótico, situando al paciente en grupos de bajo riesgo -edad inferior a los 60 años, ausencia de factores de riesgo vascular, ausencia de historia de trombosis y cifra de plaquetas inferior a 1.500 X109/l-, que no precisan tratamiento, o de alto riesgo -edad ?60 años o historia previa de trombosis- en los que la hidroxiurea asociada a AAS es el tratamiento de elección. La anagrelida evita la agregación de las plaquetas e inhibe la maduración de megacariocitos, disminuyendo de ese modo los recuentos de plaquetas. Tiene un efecto mínimo en la producción de eritrocitos, es eficaz en la reducción de recuento de plaquetas en el 70% de los pacientes, y no parece aumentar el riesgo de leucemia aguda. Sin embargo, la hidroxiurea es más eficaz que la anagrelida. Es por lo tanto, un tratamiento útil para los pacientes con trombocitosis refractarios a otras terapias.
Mielofibrosis primaria
Es una NMP caracterizada por proliferación en MO de megacariocitos y granulocitos y que posteriormente se asocia con fibrosis y hematopoyesis extramedular ?metaplasia mieloide-, y puede evolucionar a osteoesclerosis. Su incidencia anual se estima en 0,4-0,9 por cada 100,000 habitantes. Clínicamente se caracteriza por su curso insidioso, con anemia progresiva, marcada hepatoesplenomegalia y sintomatología constitucional debida al estado hipercatabólico. También son frecuentes las manifestaciones hemorrágicas debidas a trombopenia o trombopatía, complicaciones trombóticas, prurito, y en estadios avanzados la presencia de hipertensión portal, como consecuencia de la existencia de metaplasia mieloide hepática. (20)El diagnostico se basa en los siguientes criterios, requiriéndose el cumplimiento de los tres criterios mayores y al menos 2 menores:
? Criterios Mayores
1. Hiperplasia de megacariocitos atípicos, acompañados de fibrosis (reticulina/colágeno). En ausencia de fibrosis hay proliferación de granulocitos y eritropoyesis normal o disminuída.
2. Ausencia de criterios de LCM Ph+, PV, otra neoplasia mieloide ó síndrome mielodisplasico.
3. JAK 2 V617F -presente en el 50% de los casos de mielofibrosis- u otro marcador clonal (MPL W515K/L). En ausencia de estos marcadores, falta de evidencia de infección, enfermedad autoinmune u otra neoplasia.
? Criterios Menores
1. Leucoeritroblastosis
2. Incremento de LDH.
3. Anemia
4. Esplenomegalia
Los tratamientos convencionales incluyen los andrógenos como danazol, corticoides y EPO, dirigidos a paliar la sintomatología de la anemia. En las formas proliferativas son de elección los agentes citotóxicos, siendo la hidroxiurea el fármaco de elección. También la talidomida se está convirtiendo en una terapia cada vez más popular en el tratamiento de la la mielofibrosis primaria. Como alternativas pueden utilizarse interferón-?, melfalán, y busulfán, pero su uso está limitado por la baja eficacia y los efectos adversos. Para el control de la trombocitosis puede estar indicada la anagrelida. La esplenectomía y la irradiación esplénica pueden estar indicadas en casos seleccionados.
Otras NMP
En la tabla 1 se recogen las características de otras NMP menos frecuentes.
Leucemia Crónica Neutrofílica
Leucemia Crónica Eosinofílica
Mastocitosis
Neoplasia Mieloproliferativa no clasificable
Es una NMP muy poco frecuente caracterizada neutrofilia mantenida en SP, proliferación
granulocítica a expensas
de neutrófilos en MO y hepatoesplenomegalia.
En un tercio
de los pacientes se detectan alteraciones
citogenéticas como 20q-, 21+, 11q-, 12p-. Por definición el cromosoma Ph
esta ausente.
Se dispone de escasas evidencias sobre la prevalencia de la mutacion
de JAK2.
Es una NMP con proliferación clonal de precursores
eosinofílicos que ocasiona eosinofilia, en SP y MO, con infiltración de tejidos y órganos, que son dañados por citocinas originadas en los eosinófilos.
evidencia de leucemia aguda mieloblástica
Es una proliferación clonal y neoplásica de mastocitos que se acumulan en uno o más tejidos, órganos, aparatos, o sistemas. Sus distintas variantes y las alteraciones clínicas asociadas, dependen del órgano infiltrado. Hay
tres variedades: 1) Mastositosis cutánea, con infiltración confinada a la piel y de curso benigno; 2) Mastocitosis sistémica, con infiltración a piel, médula ósea y otros órganos con evolución de indolente a agresiva y que incluye
la leucemia mastocítica; 3) Mastocitosis sistémica
asociada a una alteración clonal en alguna línea celular hematológica distinta.
Esta designación es aplicable a casos con datos clínicos
y hematológicos bien definidos, como propios de NMP, pero que no reúna el criterio diagnóstico de ninguno de de ellos
ó que cumplan el de más de uno. La existencia de Ph+,
BCR-ABL1, PDGFRA, PDGB ó FGR1 excluye el diagnóstico
de NMPNC.
Tabla 1.- Características de otras Neoplasias Mieloproliferativas menos frecuentesBibliografía recomendada
Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. 2009;27(35):6041-51.
Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761-70.
Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761-70.
Barbui T, Finazzi MC, Finazzi G. Front-line therapy in polycythemia vera and essential thrombocythemia. Blood Rev. 2012; 26:205-11
Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22(2):437-8.
Begna KH, Pardanani A, Mesa R, Litzow MR, Hogan WJ, Hanson CA, Tefferi A. Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012;87:66-8.
Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. A New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895-901.
Cortelazzo S, Finazzi G, Ruggeri M, Vestri O, Galli M, Rodeghiero F, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332:1132-6.
Druker BJ, Guilhot F, O?Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355: 2408-17
González García C, Funes Vera C, Blanquer Blanquer M y Moraleda Jiménez JM. Síndromes mieloproliferativos. Medicine. 2012;11:1289-97
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787-98.
Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353:33-45.
Kantarjian HM, Shah NP, Cortes JE, Baccarani M, Agarwal MB, Undurraga MS, et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2012;119(5):1123-9.
Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673-83.
Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224-32
Moraleda JM, Hernández F. Síndromes mieloproliferativos crónicos. Leucemia mieloide crónica. En: Moraleda JM, editor. Pregrado de hematología. 3ª ed. Madrid: Luzán 5, S.A.; 2011. p. 237-56.
Najean Y, Rain JD. The very long-term evolution of polycythemia vera: an analysis of 318 patients initially treated by phlebotomy or 32P between 1969 and 1981. Semin Hematol 1997; 34:6-16.
Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus Imatinib for newly diagnosed chronic myeloid leukemia. N Eng J Med. 2010;362(24):2251-9.
Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA, Barosi G, Verstovsek S, Birgegard G, Mesa R, Reilly JT, Gisslinger H, Vannucchi AM, Cervantes F, Finazzi G, Hoffman R, Gilliland DG, Bloomfield CD, Vardiman JW. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007; 110:1092-7.
Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer 2009; 115:3842.
Tefferi A. The history of myeloproliferative disorders: before and after Dameshek. Leukemia 2008; 22:3-13.
Vannucchi AM, Guglielmelli P, Tefferi A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin 2009; 59:171-191.
Vardiman J, Hyjek E. World health organization classification, evaluation, and genetics of the myeloproliferative neoplasm variants. Hematology Am Soc Hematol Educ Program. 2011;2011:250-6.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799-807.
¿Vulnera este post tus derechos? Pincha aquí.
Modificado: