La Dra. Inmaculada Pitarch, neuropediatra, coordinadora del área pediátrica de la Unidad de Enfermedades Neuromusculares Raras del Hospital de la Fe, en Valencia, ha puesto de manifiesto la necesidad de un diagnóstico precoz para la AME en el marco de las VII Jornadas Neuropediátricas que se han desarrollado en este hospital valenciano dirigidas a neuropediatras, pediatras, médicos de Atención Primaria, médicos rehabilitadores y fisioterapeutas.
Esta especialista, que ha centrado su intervención en el diagnóstico diferencial de la debilidad muscular en la edad pediátrica, destaca que las enfermedades neuromusculares son crónicas, la mayoría de ellas comienzan a manifestarse en la infancia y tienen un componente genético y de carácter progresivo que provoca pérdida de fuerza muscular. “Son enfermedades raras, de baja prevalencia, en las que es muy importante el diagnóstico precoz para su abordaje multidisciplinar y para la instauración del tratamiento que cambiará el curso natural de la enfermedad”, subraya.
La neuropediatra apunta que las patologías que se incluyen en el cribado conocido como prueba del talón están dirigidos a enfermedades graves, con una baja prevalencia y que cuentan con tratamiento. “Actualmente, la AME es una patología que cumple con los criterios que se precisan para ser incluida en el cribado neonatal. Además, la experiencia clínica ha corroborado el gran beneficio del tratamiento precoz”, asegura.
Experiencias piloto en España
El Hospital de La Fe, además de otros centros de Andalucía y Cataluña, están desarrollando estudios piloto para la incorporación de la AME en los programas de cribado neonatal. El screening para esta patología es además una realidad en varios países en todo el mundo como Estados Unidos, Australia, Taiwán, Alemania o Bélgica, donde, tras su implementación, se han conseguido resultados excelentes en cuanto al diagnóstico, tratamiento y pronóstico de esta patología. “Esperamos que el estudio piloto de la Comunidad Valenciana sirva para la generalización a todo el Estado de esta patología en las pruebas de cribado neonatal”, comenta. “Es importante frenar la enfermedad en un paciente evolucionado, pero el gran reto para los clínicos es impedir que el niño desarrolle la patología y hoy estamos en condiciones de poder hacerlo”, añade.
Hasta 400 familias afectadas en España
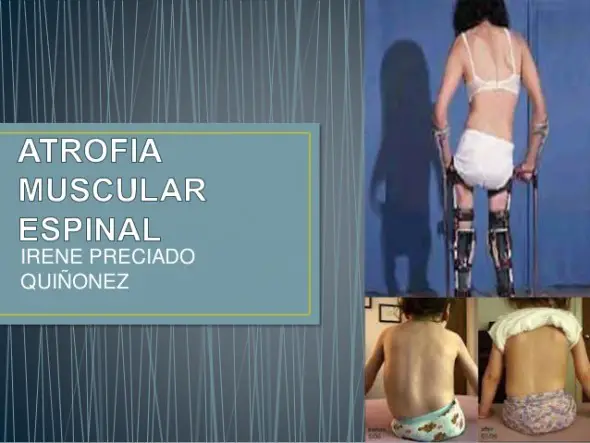
La Atrofia Muscular Espinal (AME) es una enfermedad neuromuscular de origen genético que se caracteriza por la atrofia progresiva de la musculatura que conlleva una pérdida de la fuerza y el tono muscular. Está causada por la ausencia o la anomalía del gen SMN1, lo que provoca en los pacientes una pérdida rápida e irreversible de las motoneuronas de la médula espinal con afectación de todas las funciones musculares, incluidas la respiración, la deglución y el movimiento[1],[2].La AME afecta a aproximadamente 1 de cada 10.000 nacidos vivos[3] y una de cada 54 personas es portadora de la anomalía genética[4]. Se estima que, en España entre 300 y 400 familias cuentan con algún miembro afectado. En el tipo 1, el más grave de esta enfermedad, la degeneración y pérdida de las motoneuronas comienza antes de nacer y progresa rápidamente hasta el punto de que a los 6 meses de edad se ha producido ya la pérdida de más del 95%2. Los niños que no reciben tratamiento temprano padecen una gran dependencia y discapacidad que les impedirá realizar acciones tan básicas como sentarse sin ayuda. Nueve de cada diez pacientes con AME tipo 1 mueren si no reciben tratamiento o necesitan ventilación mecánica permanente antes de los 2 años de vida[5].
En un futuro próximo los pacientes con AME dispondrán de nuevas opciones terapéuticas, que se suman a las actuales, como la terapia génica que puede frenar la evolución de la enfermedad.
--
[1] National Organization for Rare Disorders (NORD). Spinal Muscular Atrophy. http://rarediseases.org/rarediseases/spinal-muscular-atrophy/.
Accessed October 9, 2018.
[2] Anderton RS and Mastaglia FL. Advances and challenges in developing a therapy for spinal muscular atrophy. Expert Rev Neurother. 2015;15(8):895-908.
[3] National Organization for Rare Disorders (NORD). Spinal Muscular Atrophy. http://rarediseases.org/rarediseases/spinal-muscular-atrophy/. Accessed October 9, 2018.
[4] Mendell JR, et al. Single-Dose Gene Replacement Therapy for Spinal Muscular Atrophy. New Eng J Med. 2017;377(18)1713-1722.
[5] Finkel RS, McDermott MP, Kaufmann P. et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83(9):810-7.