El carcinoma de tiroides es recuente en la adultez; la incidencia anual en los niños menores de 15 años de edad es de alrededor de 2 casos/100.000, comparada con una incidencia mundial anual en todas las edades entre 4 y 10 casos/100.000. A diferencia de otras neoplasias malignas en la infancia, el cáncer de tiroides suele tener un curso insidioso en los niños, incluso después de que se hayan desarrollado metástasis pulmonares.
Causas del cáncer de tiroides
Los factores genéticos y la exposición a la radiación son factores importantes en la patogenia del cáncer de tiroides. Se encuentran reordenamientos del protooncogén RET en un 3-33% de los carcinomas papilares y en un 60-80% de los que se producen tras irradiación, como en los niños de Bielorrusia expuestos a la radiación tras el accidente nuclear de Chernóbil o en los que estuvieron expuestos a una irradiación externa terapéutica durante la infancia. Las mutaciones puntuales inactivadoras del gen supresor tumoral p53 son infrecuentes en los pacientes con carcinoma diferenciado de tiroides, pero son habituales en aquellos con carcinoma anaplásico de tiroides. En conjunto, el 5-10% de los casos de carcinoma papilar de tiroides son familiares y se heredan generalmente mediante herencia autosómica dominante.

La glándula tiroides de los niños es extraordinariamente sensible a la exposición a la radiación externa. Es probable que no exista dosis umbral; 1 Gy produce un riesgo relativo de 7,7 de cáncer de tiroides. En el pasado, alrededor del 80% de los niños con cáncer de tiroides había recibido radioterapia en el cuello y las áreas adyacentes durante la lactancia para tratar enfermedades benignas como el timo «de gran tamaño », amígdalas y adenoides hipertróficas, hemangiomas, nevos, dermatitis, tiña de la cabeza y «adenitis cervical». Desde que no se emplea la radioterapia para entidades benignas, esta causa de cáncer tiroideo ha desaparecido.
Tipos de cáncer de tiroides
Histológicamente, los carcinomas de tiroides son papilares (80%), foliculares (17%), medulares (2%) o tumores mixtos diferenciados. Suelen ser tumores de crecimiento lento y pueden permanecer silentes durante años.
El tipo de tumor y el curso natural de la enfermedad en los pacientes irradiados y en los no irradiados son iguales, y sólo se diferencian porque la multicentricidad es más frecuente en el cáncer inducido por radioterapia. Las neoplasias tiroideas indiferenciadas (anaplásicas) son infrecuentes en los niños y tienen habitualmente un curso rápido y mortal. El cáncer de tiroides, sobre todo el de tipo papilar, se ha descrito en niños con restos del conducto tirogloso. También se han descrito casos pediátricos mde linfomas y teratomas de la glándula tiroides.
1. Manifestaciones clínicas
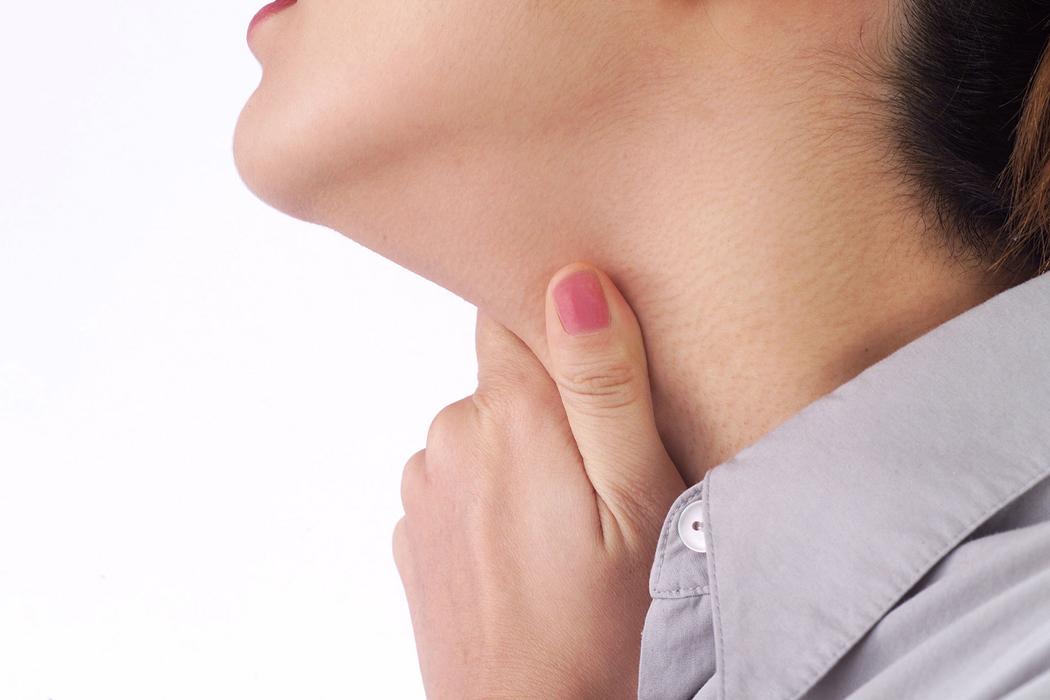
Las niñas se afectan dos veces más que los niños de carcinoma de tiroides. La edad media en el momento del diagnóstico es de 9 años, pero puede presentarse incluso en el primer año de vida. El primer signo de la enfermedad suele ser un nódulo tiroideo o cervical no doloroso. La presencia de un nódulo grande, firme y fijado a los tejidos adyacentes, así como la parálisis de la cuerda vocal, son factores de riesgo de cáncer de tiroides. La afectación ganglionar cervical suele estar presente en el momento del diagnóstico inicial.
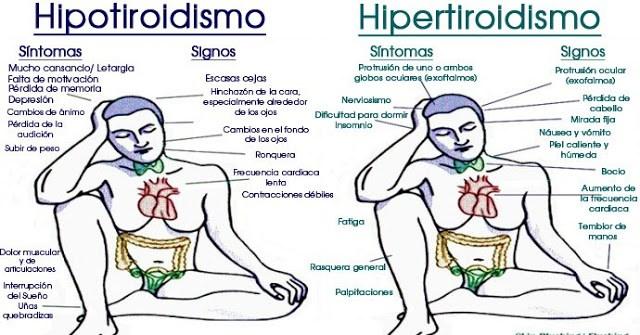
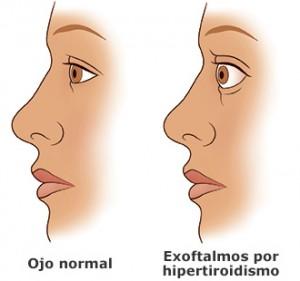
Cualquier adenopatía cervical no explicada requiere una exploración de la glándula tiroides, que en ocasiones tiene un tumor primario demasiado pequeño para ser palpado; el diagnóstico se basa en los resultados de la biopsia del ganglio linfático. Los pulmones son el lugar más frecuente de metástasis fuera del cuello. Puede no haber manifestaciones clínicas secundarias a ellas; en la radiología aparecen infiltraciones miliares difusas o nodulares, sobre todo en las bases. Pueden ser confundidas con tuberculosis, histoplasmosis o sarcoidosis. Otras localizaciones de metástasis son el mediastino, los huesos largos, el cráneo y la axila. Casi todos los niños son eutiroideos, pero en pocas ocasiones el carcinoma puede ser funcional y producir síntomas de hipertiroidismo
Tratamiento
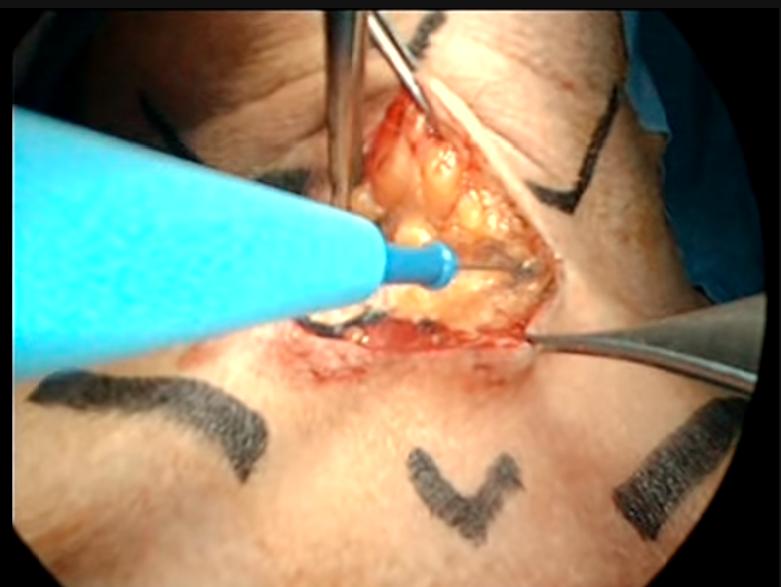
El tratamiento óptimo está todavía en desarrollo del carcinoma de tiroides, ya que el carcinoma diferenciado de tiroides es una enfermedad crónica con una larga supervivencia. El carcinoma papilar pequeño (<1 cm), que es el tipo menos agresivo, se trata con eficacia mediante tiroidectomía subtotal y dosis supresoras de hormona tiroidea. Sin embargo, existen cada vez más datos de que en la mayoría de los pacientes el mejor tratamiento es la tiroidectomía total o casi total. Los carcinomas papilares tienden a ser multicéntricos y diversos estudios muestran que la mitad de estos niños tienen afectación ganglionar regional en el momento del diagnóstico.
En los carcinomas papilares de mayor tamaño, los carcinomas foliculares o con afectación ganglionar regional, el tratamiento de elección es la tiroidectomía total con resección de los ganglios linfáticos regionales.
No procede una disección cervical radical. La tiroidectomía suele seguirse de una dosis de ablación (30-100 mCi) . Sólo los pacientes sometidos a tiroidectomía total pueden controlarse mediante gammagrafía corporal total con yodo radiactivo y con medición de la tiroglobulina plasmática.
Pronostico
Para cualquier modalidad de tratamiento de carcinoma de tiroides, la supervivencia o la recidiva no parecen ser diferentes en los pacientes con o sin afectación de los ganglios linfáticos cervicales. Incluso pacientes con metástasis cervicales o pulmonares han sobrevivido muchos años. Más del 95% de los pacientes está vivo 25 años después del tratamiento inicial si el tumor era intratiroideo, menor de 2 cm de diámetro y clasificado como de grado 1. Un tamaño tumoral mayor, la diseminación a distancia y una atipia de mayor grado se asocian con mayor mortalidad acumulada. Los carcinomas tiroideos que expresan telomerasa y el receptor de IGF-1 tienen mayor probabilidad de presentar características clínicas agresivas, mientras que los que expresan el cotransportador de sodio-yoduro se asocian a un menor riesgo de recidiva

Nódulo Tiroideo Solitario
Los nódulos tiroideos solitarios son infrecuentes en los niños. Se descubren por palpación en alrededor del 2% de los niños de 11-18 años; es probable que la exploración mediante ecografía mostrase una mayor prevalencia, como sucede en adultos. Los factores genéticos intervienen en la etiología. La mayoría de los nódulos tiroideos son tumores benignos. La incidencia de malignidad es de alrededor del 15%, quizá por la menor exposición de los niños a la radiación. Los niños expuestos a la radiación tienen una alta incidencia de adenoma benigno y de carcinoma tiroideo.
Los trastornos benignos que pueden presentarse como nódulos tiroideos solitarios incluyen los adenomas benignos (p. ej., folicular, embrionario o de células de Hürthle), nódulos coloides (adenomatosos), quiste simple, tiroiditis linfocitaria, absceso tiroideo y anomalías del desarrollo, tales como quiste del conducto tirogloso o hemiagenesia. Una masa tiroidea que aparece de forma brusca o que aumenta de tamaño también pueden calcificarse. Puede producir la muerte, pero lo habitual es una supervivencia larga.
El carcinoma de tiroides se presentan de forma esporádica, como un trastorno familiar autosómico dominante y como componentes de dos síndromes autosómicos dominantes distintos. La susceptibilidad para todos estos trastornos se ha asociado a mutaciones en la línea germinal del protooncogén RET, localizado en el cromosoma 10q11.2. Cuando el tumor se presenta de forma esporádica es habitualmente unicéntrico, pero en la forma familiar suele ser multicéntrico y comienza como una hiperplasia de las células parafoliculares.
Los tumores del carcinoma de tiroides con frecuencia son demasiado pequeños para encontrarse mediante palpación, gammagrafía o ecografía en los pacientes de riesgo en estas familias. El diagnóstico de carcinoma medular debe promover la búsqueda cuidadosa de tumores asociados, en especial de feocromocitoma. Los niveles plasmáticos elevados de calcitonina o del péptido relacionado con el gen de la calcitonina no producen manifestaciones clínicas. Sin embargo, estas pruebas son útiles para la detección selectiva y el control del tratamiento.
Neoplasia Endrocrina Múltiple Tipo IIA
Cuando la hiperplasia o el carcinoma de células C se asocia a hiperplasia medular suprarrenal o a feocromocitoma e hiperplasia paratiroidea, se conoce como neoplasia endocrina múltiple (MEN) IIA. El patrón de herencia del MEN IIA es autosómico dominante, con un alto grado de penetrancia y expresividad variable. Se han descrito al menos 19 mutaciones de aminoácido específicas diferentes en el exón 10 o en el 11 del dominio extracelular del gen RET en la MEN IIA y en casos de carcinoma tiroideo medular familiar.
Los análisis de ADN permiten la identificación inequívoca de los portadores del protooncogén RET. La hiperplasia o los tumores de células C generalmente aparecen antes que el feocromocitoma, que suele ser bilateral y puede ser múltiple. Se sabe que la hiperplasia suprarrenal medular precede al feocromocitoma, pero el período de latencia detectable es corto. La hipercalcemia es una manifestación tardía e indica hiperparatiroidismo.
Las glándulas paratiroides pueden mostrar hiperplasia de células principales o únicamente hipercelularidad.
Neoplasia Endrocrina Múltiple Tipo IIB
La característica diferenciadora de la MEN IIB, también llamada síndrome del neuroma mucoso, es la presentación de neuromas múltiples y un fenotipo típico asociado con el carcinoma medular y el feocromocitoma. Este trastorno es también autosómico dominante, y el 93% de las familias tiene una mutación de aminoácido del protooncogén RET. Sin embargo, la mutación es en el exón 16, el dominio tirosina-cinasa de RET; todos los pacientes tienen la misma mutación puntual. Los neuromas se presentan con mayor frecuencia en la lengua, la mucosa oral, los labios y las conjuntivas.

Puede haber neurofibromas periféricos y manchas café con leche en carcinoma de tiroides, y la ganglioneuromatosis intestinal es frecuente. La proliferación difusa de nervios y células ganglionares se localiza en los plexos mucoso, submucoso, mientérico y subseroso del intestino delgado y grueso, así como del esófago. Los pacientes pueden ser altos, con aracnodactilia y un aspecto similar al síndrome de Marfan. Son frecuentes la escoliosis, el pectus excavatum, los pies cavos y la hipotonía muscular. Los párpados pueden estar engrosados y evertidos, los labios inflamados y distendidos, y la mandíbula prominente. Las dificultades en la alimentación y en la succión, la diarrea, el estreñimiento y el retraso del crecimiento pueden comenzar en la lactancia o en la fase precoz de la infancia, muchos años antes de la aparición de los neuromas o los síntomas endocrinos.
Tratamiento
La tiroidectomía total está indicada en todos los niños portadores del gen demostrado mediante estudios de ADN. El reconocimiento de las formas familiares de este tumor es fundamental para el diagnóstico precoz de los niños con riesgo. Las pruebas sugieren que la tiroidectomía debe realizarse lo antes posible, porque se ha encontrado un carcinoma medular en un niño de 6 meses de edad con MEN IIB y en un niño de 3 años de edad con MEN IIA. En el MEN IIA, existe una correlación entre la mutación específica y el inicio de la hiperplasia de células C o del carcinoma medular.
En los niños pequeños con carcinoma de tiroides, las mutaciones en el codón 634 se producen a una edad temprana, mientras que las de los codones 618, 620 y 804 tienden a producirse más tarde. En todos estos niños se debería realizar una detección selectiva del feocromocitoma antes de la cirugía. La monitorización de los niveles de calcitonina es útil en el seguimiento evolutivo de la enfermedad tras la intervención y en la detección de lesiones metastásicas. Está indicado el cribado periódico para detectar el desarrollo del feocromocitoma y el hiperparatiroidismo.
.